
The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
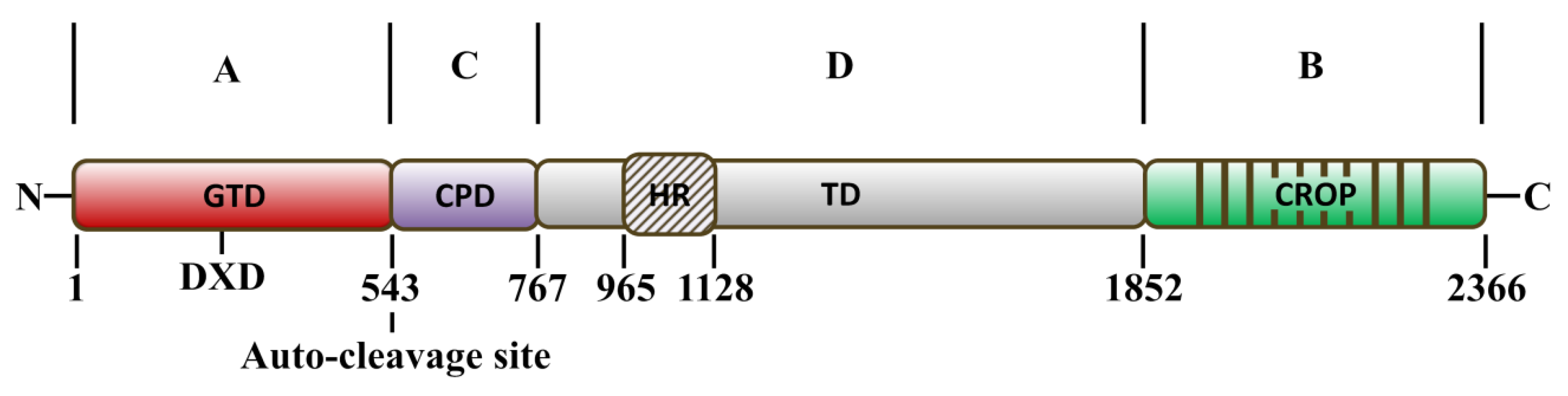
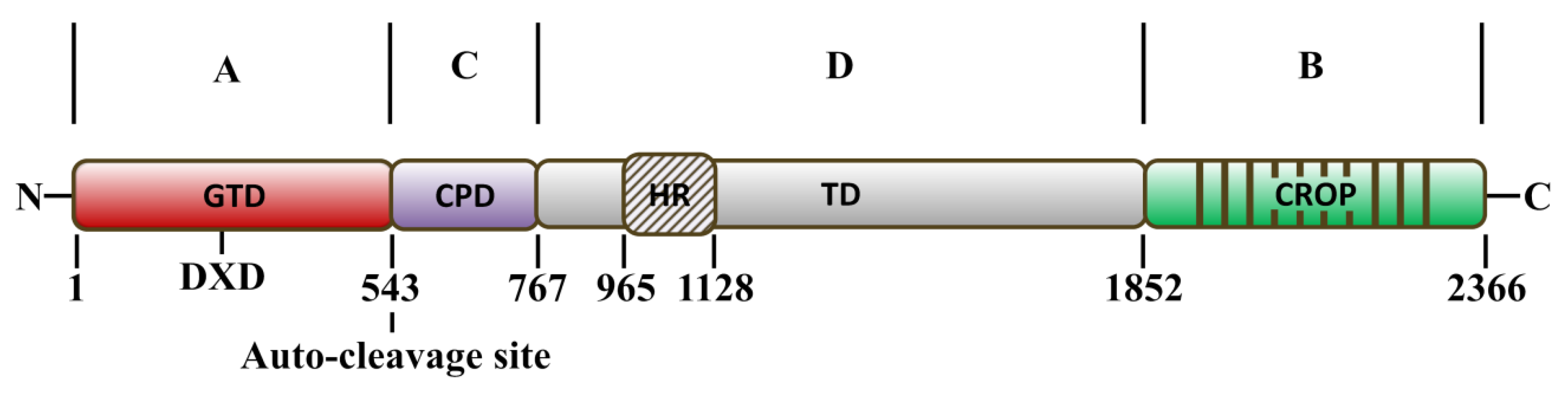
2. Structure–Function Relationship and Mechanism of TcdA and TcdB
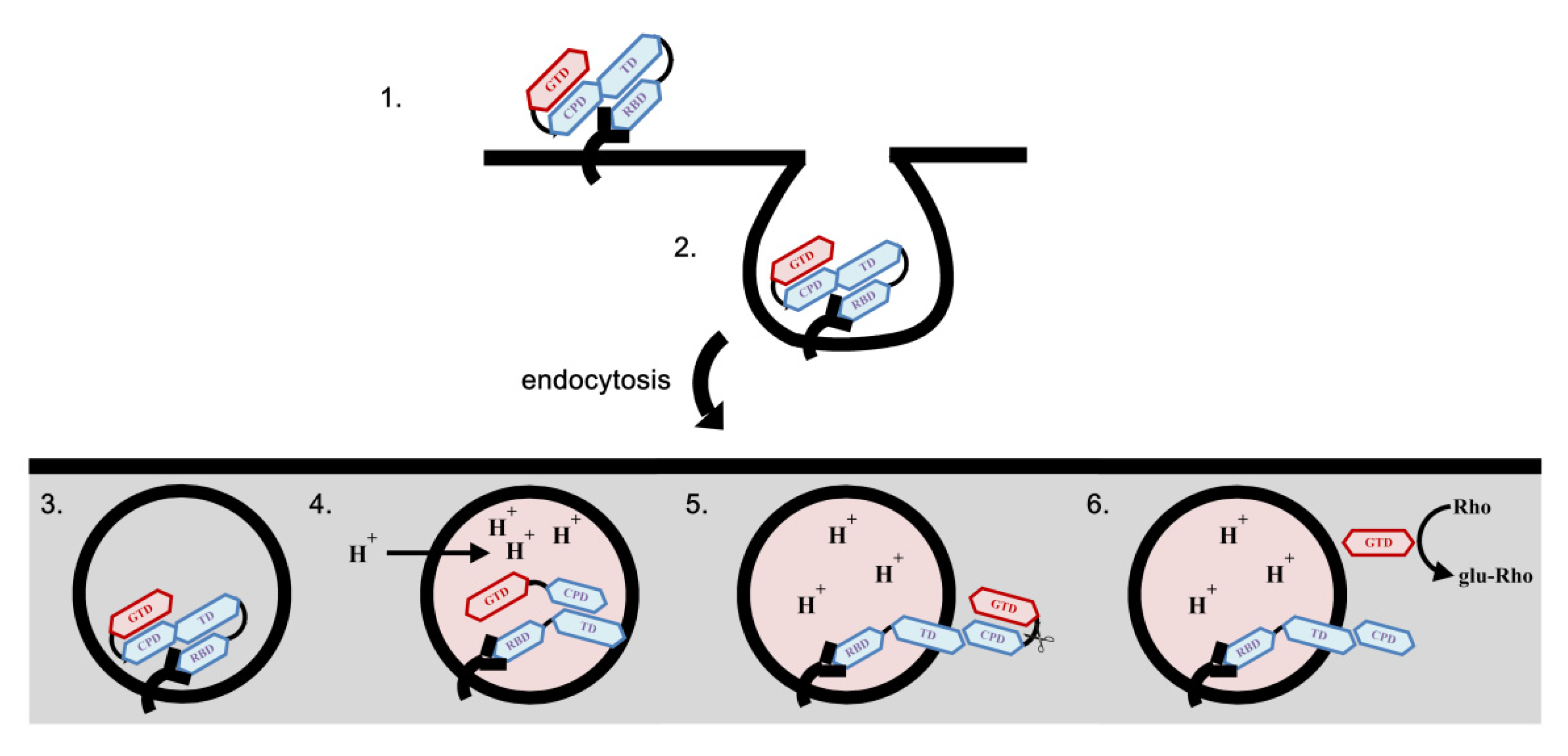
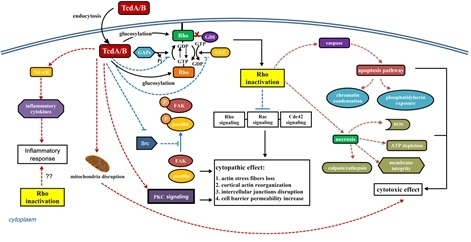
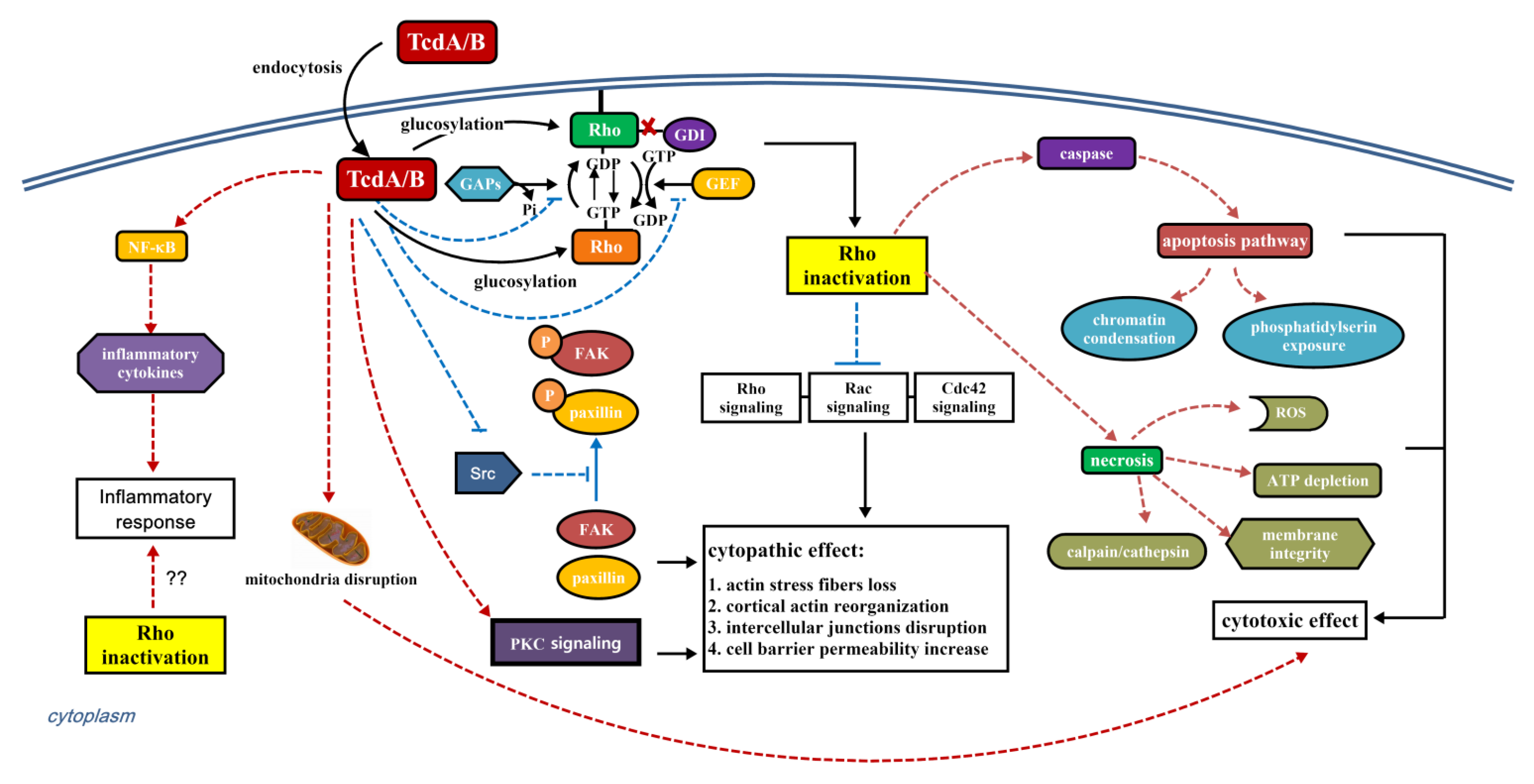
3. Interaction of C. difficile Toxins and Rho GTPases
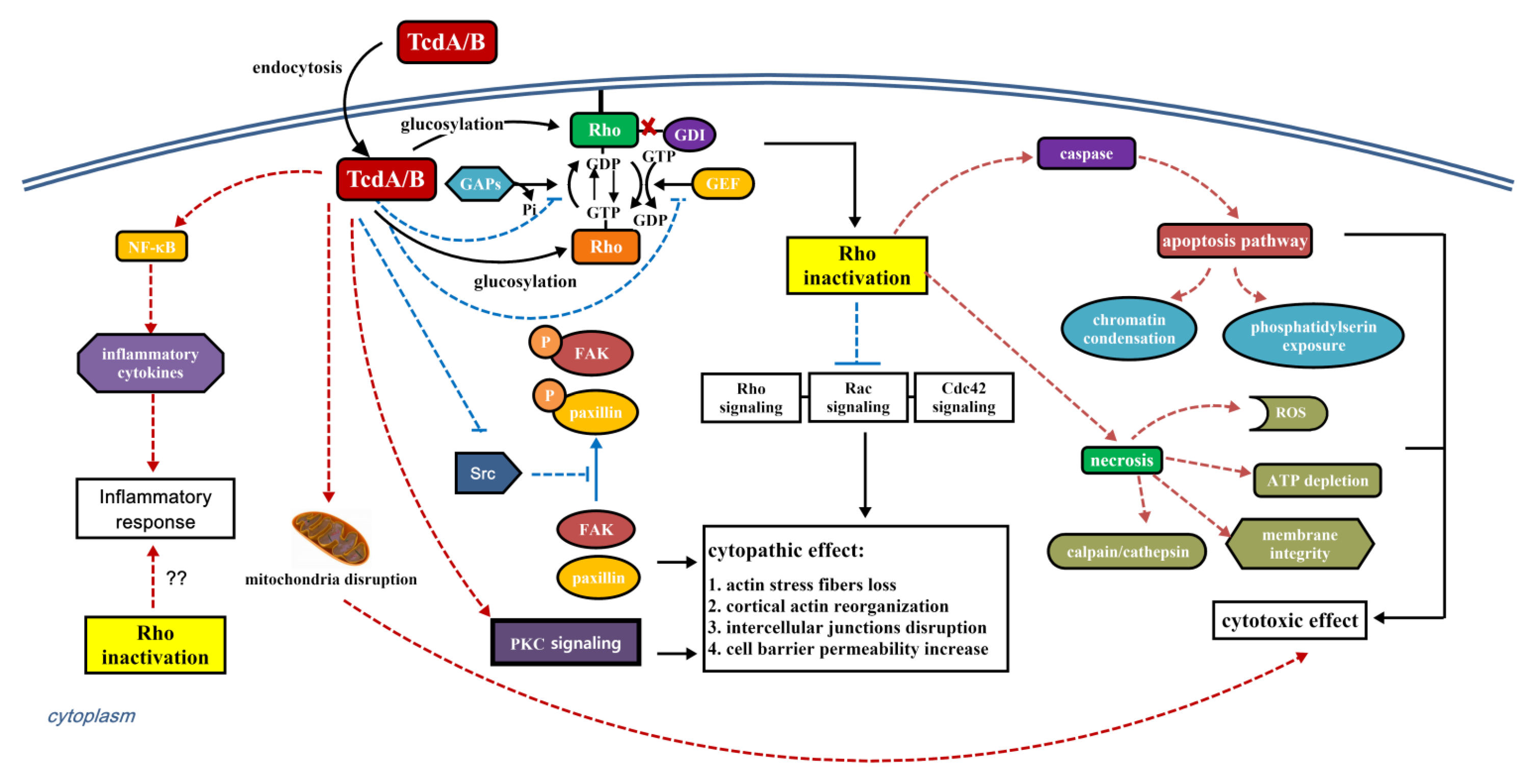
4. The Role of Rho GTPase Glucosylation in Toxicity of TcdA and TcdB
4.1. Cytopathic and Cytotoxic Effect
4.2. Immune Response
5. Conclusions and Future Considerations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartlett, J.G.; Chang, T.W.; Gurwith, M.; Gorbach, S.L.; Onderdonk, A.B. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N. Engl. J. Med. 1978, 298, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Planche, T.; Wilcox, M.H. Diagnostic pitfalls in Clostridium difficile infection. Infect. Dis. Clin. N. Am. 2015, 29, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Luciano, J.A.; Zuckerbraun, B.S. Clostridium difficile infection: Prevention, treatment, and surgical management. Surg. Clin. N. Am. 2014, 94, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Bagdasarian, N.; Rao, K.; Malani, P.N. Diagnosis and treatment of Clostridium difficile in adults: A systematic review. JAMA 2015, 313, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Pepin, J.; Alary, M.E.; Valiquette, L.; Raiche, E.; Ruel, J.; Fulop, K.; Godin, D.; Bourassa, C. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2005, 40, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Polyzos, K.A.; Patouni, K.; Rafailidis, P.I.; Samonis, G.; Falagas, M.E. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: A systematic review of the evidence. Int. J. Antimicrob. Agents 2012, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shields, K.; Araujo-Castillo, R.V.; Theethira, T.G.; Alonso, C.D.; Kelly, C.P. Recurrent Clostridium difficile infection: From colonization to cure. Anaerobe 2015, 34, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Zimlichman, E.; Henderson, D.; Tamir, O.; Franz, C.; Song, P.; Yamin, C.K.; Keohane, C.; Denham, C.R.; Bates, D.W. Health care-associated infections: A meta-analysis of costs and financial impact on the US health care system. JAMA Intern. Med. 2013, 173, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile infection. Ann. Rev. Med. 1998, 49, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Gerhard, R. Large clostridial cytotoxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 23–47. [Google Scholar] [PubMed]
- Von Eichel-Streiber, C.; Boquet, P.; Sauerborn, M.; Thelestam, M. Large clostridial cytotoxins—A family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol. 1996, 4, 375–382. [Google Scholar] [CrossRef]
- Schirmer, J.; Aktories, K. Large clostridial cytotoxins: Cellular biology of Rho/Ras-glucosylating toxins. Biochim. Biophys. Acta 2004, 1673, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Wilm, M.; Selzer, J.; Rex, G.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J. Biol. Chem. 1995, 270, 13932–13936. [Google Scholar] [CrossRef]
- Just, I.; Selzer, J.; Hofmann, F.; Green, G.A.; Aktories, K. Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J. Biol. Chem. 1996, 271, 10149–10153. [Google Scholar]
- Popoff, M.R.; Chaves-Olarte, E.; Lemichez, E.; von Eichel-Streiber, C.; Thelestam, M.; Chardin, P.; Cussac, D.; Antonny, B.; Chavrier, P.; Flatau, G.; et al. Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J. Biol. Chem. 1996, 271, 10217–10224. [Google Scholar]
- Selzer, J.; Hofmann, F.; Rex, G.; Wilm, M.; Mann, M.; Just, I.; Aktories, K. Clostridium novyi alpha-toxin-catalyzed incorporation of GlcNAc into Rho subfamily proteins. J. Biol. Chem. 1996, 271, 25173–25177. [Google Scholar] [CrossRef]
- Ottlinger, M.E.; Lin, S. Clostridium difficile toxin B induces reorganization of actin, vinculin, and talin in cultured cells. Exp. Cell Res. 1988, 174, 215–229. [Google Scholar] [CrossRef]
- Prepens, U.; Just, I.; von Eichel-Streiber, C.; Aktories, K. Inhibition of Fc epsilon-RI-mediated activation of rat basophilic leukemia cells by Clostridium difficile toxin B (monoglucosyltransferase). J. Biol. Chem. 1996, 271, 7324–7329. [Google Scholar]
- Schmidt, M.; Rumenapp, U.; Bienek, C.; Keller, J.; von Eichel-Streiber, C.; Jakobs, K.H. Inhibition of receptor signaling to phospholipase D by Clostridium difficile toxin B. Role of Rho proteins. J. Biol. Chem. 1996, 271, 2422–2426. [Google Scholar] [CrossRef]
- Subauste, M.C.; von Herrath, M.; Benard, V.; Chamberlain, C.E.; Chuang, T.H.; Chu, K.; Bokoch, G.M.; Hahn, K.M. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J. Biol. Chem. 2000, 275, 9725–9733. [Google Scholar] [CrossRef]
- Caron, E.; Hall, A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 1998, 282, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: The ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Von Eichel-Streiber, C.; Sauerborn, M.; Kuramitsu, H.K. Evidence for a modular structure of the homologous repetitive C-terminal carbohydrate-binding sites of Clostridium difficile toxins and Streptococcus mutans glucosyltransferases. J. Bacteriol. 1992, 174, 6707–6710. [Google Scholar]
- Barroso, L.A.; Moncrief, J.S.; Lyerly, D.M.; Wilkins, T.D. Mutagenesis of the Clostridium difficile toxin B gene and effect on cytotoxic activity. Microb. Pathog. 1994, 16, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Hofmann, F.; Selzer, J.; Munro, S.; Jeckel, D.; Aktories, K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J. Biol. Chem. 1998, 273, 19566–19572. [Google Scholar] [CrossRef]
- Jank, T.; Giesemann, T.; Aktories, K. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 2007, 282, 35222–35231. [Google Scholar] [CrossRef] [PubMed]
- Reineke, J.; Tenzer, S.; Rupnik, M.; Koschinski, A.; Hasselmayer, O.; Schrattenholz, A.; Schild, H.; von Eichel-Streiber, C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007, 446, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Egerer, M.; Giesemann, T.; Jank, T.; Satchell, K.J.; Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007, 282, 25314–25321. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, R.N.; Chagot, B.; Cover, M.; Chazin, W.J.; Spiller, B.; Lacy, D.B. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009, 284, 21934–21940. [Google Scholar] [CrossRef] [PubMed]
- Kreimeyer, I.; Euler, F.; Marckscheffel, A.; Tatge, H.; Pich, A.; Olling, A.; Schwarz, J.; Just, I.; Gerhard, R. Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Von Eichel-Streiber, C.; Sauerborn, M. Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 1990, 96, 107–113. [Google Scholar] [CrossRef]
- Dove, C.H.; Wang, S.Z.; Price, S.B.; Phelps, C.J.; Lyerly, D.M.; Wilkins, T.D.; Johnson, J.L. Molecular characterization of the Clostridium difficile toxin A gene. Infect. Immun. 1990, 58, 480–488. [Google Scholar] [PubMed]
- Moncrief, J.S.; Wilkins, T.D. Genetics of Clostridium difficile toxins. Curr. Top. Microbiol. Immunol. 2000, 250, 35–54. [Google Scholar] [PubMed]
- Ho, J.G.; Greco, A.; Rupnik, M.; Ng, K.K. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc. Natl. Acad. Sci. USA 2005, 102, 18373–18378. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Ho, J.G.; Lin, S.J.; Palcic, M.M.; Ng, K.K. Carbohydrate recognition by Clostridium difficile toxin A. Nat. struct. Mol. Biol. 2006, 13, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Qa’Dan, M.; Spyres, L.M.; Ballard, J.D. pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 2000, 68, 2470–2474. [Google Scholar] [CrossRef]
- Barth, H.; Pfeifer, G.; Hofmann, F.; Maier, E.; Benz, R.; Aktories, K. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J. Biol. Chem. 2001, 276, 10670–10676. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.; Schirmer, J.; Leemhuis, J.; Busch, C.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 2003, 278, 44535–44541. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.O.; Jank, T.; Aktories, K.; Schulz, G.E. Conformational changes and reaction of clostridial glycosylating toxins. J. Mol. Biol. 2008, 377, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Genisyuerek, S.; Papatheodorou, P.; Guttenberg, G.; Schubert, R.; Benz, R.; Aktories, K. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. 2011, 79, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Rupnik, M.; Pabst, S.; Rupnik, M.; von Eichel-Streiber, C.; Urlaub, H.; Soling, H.D. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 2005, 151, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Florin, I.; Thelestam, M. Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochim. Biophys. Acta 1983, 763, 383–392. [Google Scholar] [CrossRef]
- Henrigues, B.; Florin, I.; Thelestam, M. Cellular internalisation of Clostridium difficile toxin A. Microb. Pathog. 1987, 2, 455–463. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Jank, T.; Giesemann, T.; Aktories, K. Rho-glucosylating Clostridium difficile toxins A and B: New insights into structure and function. Glycobiology 2007, 17, 15R–22R. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Olarte, E.; Weidmann, M.; Eichel-Streiber, C.; Thelestam, M. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J. Clin. Investig. 1997, 100, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Mehlig, M.; Moos, M.; Braun, V.; Kalt, B.; Mahony, D.E.; von Eichel-Streiber, C. Variant toxin B and a functional toxin A produced by Clostridium difficile C34. FEMS Microbiol. Lett. 2001, 198, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Pauillac, S.; Schelle, I.; Bouvet, P.; Bouchier, C.; Varela-Chavez, C.; Just, I.; Popoff, M.R. Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: Molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell Microbiol. 2014, 16, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Ahmadian, M.R.; Hofmann, F.; Just, I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J. Biol. Chem. 1998, 273, 16134–16139. [Google Scholar] [CrossRef] [PubMed]
- Sehr, P.; Joseph, G.; Genth, H.; Just, I.; Pick, E.; Aktories, K. Glucosylation and ADP ribosylation of rho proteins: Effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry 1998, 37, 5296–5304. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Dreger, S.C.; Huelsenbeck, J.; Just, I. Clostridium difficile toxins: More than mere inhibitors of Rho proteins. Int. J. Biochem. Cell Biol. 2008, 40, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Reinert, D.J.; Jank, T.; Aktories, K.; Schulz, G.E. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005, 351, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Unligil, U.M.; Rini, J.M. Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol. 2000, 10, 510–517. [Google Scholar] [CrossRef]
- Qasba, P.K.; Ramakrishnan, B.; Boeggeman, E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem. Sci. 2005, 30, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Pack, U.; Giesemann, T.; Schmidt, G.; Aktories, K. Exchange of a single amino acid switches the substrate properties of RhoA and RhoD toward glucosylating and transglutaminating toxins. J. Biol. Chem. 2006, 281, 19527–19535. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; von Eichel-Streiber, C.; Moos, M. Impact of amino acids 22–27 of Rho-subfamily GTPases on glucosylation by the large clostridial cytotoxins TcsL-1522, TcdB-1470 and TcdB-8864. Eur. J. Biochem. 1999, 266, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Aktories, K.; Just, I. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J. Biol. Chem. 1999, 274, 29050–29056. [Google Scholar] [CrossRef] [PubMed]
- Kozma, R.; Ahmed, S.; Best, A.; Lim, L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 1995, 15, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Gerhard, R.; Schmidt, G.; Hofmann, F.; Aktories, K. Activation of Rho GTPases by Escherichia coli cytotoxic necrotizing factor 1 increases intestinal permeability in Caco-2 cells. Infect. Immun. 1998, 66, 5125–5131. [Google Scholar] [PubMed]
- Hecht, G.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin A perturbs cytoskeletal structure and tight juction permeability of cultured human intestinal epithelial monolayers. J. Clin. Investig. 1988, 82, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Hecht, G.; Koutsouris, A.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 1992, 102, 416–423. [Google Scholar] [PubMed]
- Johal, S.S.; Solomon, K.; Dodson, S.; Borriello, P.; Mahida, Y.R. Differential effects of varying concentrations of Clostridium difficile toxin A on epithelial barrier function and expression of cytokines. J. Infect. Dis. 2004, 189, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.; Pothoulakis, C.; LaMont, J.T.; Carlson, S.; Madara, J.L. C. difficile toxin A increases intestinal permeability and induces Cl-secretion. Am. J. Physiol. 1990, 259, G165–G172. [Google Scholar] [PubMed]
- Triadafilopoulos, G.; Pothoulakis, C.; O’Brien, M.J.; LaMont, J.T. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 1987, 93, 273–279. [Google Scholar] [PubMed]
- Triadafilopoulos, G.; Pothoulakis, C.; Weiss, R.; Tiampaolo, C.; LaMont, J.T. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Gastroenterology 1989, 97, 1186–1192. [Google Scholar] [PubMed]
- Chaves-Olarte, E.; Freer, E.; Parra, A.; Guzman-Verri, C.; Moreno, E.; Thelestam, M. R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J. Biol. Chem. 2003, 278, 7956–7963. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Pothoulakis, C.; LaMont, J.T. Protein kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile toxin A. J. Biol. Chem. 2002, 277, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Rhee, S.H.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile toxin A binds colonocyte Src causing dephosphorylation of focal adhesion kinase and paxillin. Exp. Cell Res. 2009, 315, 3336–3344. [Google Scholar] [CrossRef] [PubMed]
- Halabi-Cabezon, I.; Huelsenbeck, J.; May, M.; Ladwein, M.; Rottner, K.; Just, I.; Genth, H. Prevention of the cytopathic effect induced by Clostridium difficile Toxin B by active Rac1. FEBS Lett. 2008, 582, 3751–3756. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Dreger, S.C.; Gerhard, R.; Fritz, G.; Just, I.; Genth, H. Upregulation of the immediate early gene product RhoB by exoenzyme C3 from Clostridium limosum and toxin B from Clostridium difficile. Biochemistry 2007, 46, 4923–4931. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, R.; Tatge, H.; Genth, H.; Thum, T.; Borlak, J.; Fritz, G.; Just, I. Clostridium difficile toxin A induces expression of the stress-induced early gene product RhoB. J. Biol. Chem. 2005, 280, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Huelsenbeck, J.; Hartmann, B.; Hofmann, F.; Just, I.; Gerhard, R. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett. 2006, 580, 3565–3569. [Google Scholar] [CrossRef] [PubMed]
- Mahida, Y.R.; Galvin, A.; Makh, S.; Hyde, S.; Sanfilippo, L.; Borriello, S.P.; Sewell, H.F. Effect of Clostridium difficile Toxin A on Human Colonic Lamina Propria Cells: Early Loss of Macrophages Followed by T-Cell Apoptosis. Infect. Immun. 1998, 66, 5462–5469. [Google Scholar] [PubMed]
- Gerhard, R.; Nottrott, S.; Schoentaube, J.; Tatge, H.; Olling, A.; Just, I. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J. Med. Microbiol. 2008, 57, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Brito, G.A.; Fujji, J.; Carneiro-Filho, B.A.; Lima, A.A.; Obrig, T.; Guerrant, R.L. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J. Infect. Dis. 2002, 186, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Hippenstiel, S.; Schmeck, B.; N’Guessan, P.D.; Seybold, J.; Krull, M.; Preissner, K.; Eichel-Streiber, C.V.; Suttorp, N. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L830–L838. [Google Scholar] [CrossRef] [PubMed]
- Teichert, M.; Tatge, H.; Schoentaube, J.; Just, I.; Gerhard, R. Application of mutated Clostridium difficile toxin A for determination of glucosyltransferase-dependent effects. Infect. Immun. 2006, 74, 6006–6010. [Google Scholar] [CrossRef] [PubMed]
- Nottrott, S.; Schoentaube, J.; Genth, H.; Just, I.; Gerhard, R. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis Int. J. Program. Cell Death 2007, 12, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Dreger, S.; Gerhard, R.; Barth, H.; Just, I.; Genth, H. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect. Immun. 2007, 75, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, X.; Zhang, Y.; Li, S.; Chen, K.; Shi, L.; Nie, W.; Kumar, R.; Tzipori, S.; Wang, J.; et al. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect. Immun. 2012, 80, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, H.; Chen, S.; Li, Z.; Li, S.; Wang, J. Recombinant Clostridium difficile toxin B induces endoplasmic reticulum stress in mouse colonal carcinoma cells. Acta Biochim. Biophys. Sin. 2014, 46, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Chumbler, N.M.; Farrow, M.A.; Lapierre, L.A.; Franklin, J.L.; Haslam, D.B.; Goldenring, J.R.; Lacy, D.B. Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog. 2012. [Google Scholar] [CrossRef]
- Jafari, N.V.; Kuehne, S.A.; Bryant, C.E.; Elawad, M.; Wren, B.W.; Minton, N.P.; Allan, E.; Bajaj-Elliott, M. Clostridium difficile modulates host innate immunity via toxin-independent and dependent mechanism(s). PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Solomon, K. The host immune response to Clostridium difficile infection. Ther. Adv. Infect. Dis. 2013, 1, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile colitis. N. Engl. J. Med. 1994, 330, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Hirota, S.A.; Gross, O.; Li, Y.; Ulke-Lemee, A.; Potentier, M.S.; Schenck, L.P.; Vilaysane, A.; Seamone, M.E.; Feng, H.; et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 2010, 139, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Warny, M.; Keates, A.C.; Keates, S.; Castagliuolo, I.; Zacks, J.K.; Aboudola, S.; Qamar, A.; Pothoulakis, C.; LaMont, J.T.; Kelly, C.P. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J. Clin. Investig. 2000, 105, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.Y.; Lin, C.N.; Chang, C.F.; Lin, C.H.; Lien, H.T.; Chen, J.Y.; Chia, J.S. C-terminal repeats of Clostridium difficile toxin A induce production of chemokine and adhesion molecules in endothelial cells and promote migration of leukocytes. Infect. Immun. 2008, 76, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, X.; Tzipori, S.; Gerhard, R.; Feng, H. Essential role of the glucosyltransferase activity in Clostridium difficile toxin-induced secretion of TNF-alpha by macrophages. Microb. Pathog. 2009, 46, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Castagliuolo, I.; Keates, A.C.; Wang, C.C.; Pasha, A.; Valenick, L.; Kelly, C.P.; Nikulasson, S.T.; LaMont, J.T.; Pothoulakis, C. Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J. Immunol. 1998, 160, 6039–6045. [Google Scholar] [PubMed]
- Kim, J.M.; Lee, J.Y.; Yoon, Y.M.; Oh, Y.K.; Youn, J.; Kim, Y.J. NF-kappa B activation pathway is essential for the chemokine expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. Scand. J. Immunol. 2006, 63, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, K.K.; Smith, M.F., Jr.; Bobak, D.A. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J. Immunol. 1999, 163, 5183–5191. [Google Scholar] [PubMed]
- Yoshino, Y.; Kitazawa, T.; Ikeda, M.; Tatsuno, K.; Yanagimoto, S.; Okugawa, S.; Yotsuyanagi, H.; Ota, Y. Clostridium difficile flagellin stimulates toll-like receptor 5, and toxin B promotes flagellin-induced chemokine production via TLR5. Life Sci. 2013, 92, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Li, S.; Li, G.; Tian, Y.; Wang, H.; Shi, L.; Perez-Cordon, G.; Mao, L.; Wang, X.; Wang, J.; et al. Utility of Clostridium difficile toxin B for inducing anti-tumor immunity. PLoS ONE 2014, 9, e110826. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Kaibuchi, K.; Ando, S.; Musha, T.; Hiraoka, K.; Takaishi, K.; Asada, M.; Nunoi, H.; Matsuda, I.; Takai, Y. Regulation of the superoxide-generating NADPH oxidase by a small GTP-binding protein and its stimulatory and inhibitory GDP/GTP exchange proteins. J. Biol. Chem. 1992, 267, 10215–10218. [Google Scholar] [PubMed]
- Heyworth, P.G.; Knaus, U.G.; Xu, X.; Uhlinger, D.J.; Conroy, L.; Bokoch, G.M.; Curnutte, J.T. Requirement for posttranslational processing of Rac GTP-binding proteins for activation of human neutrophil NADPH oxidase. Mol. Biol. Cell 1993, 4, 261–269. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Sulciner, D.J.; Gutkind, J.S.; Irani, K.; Goldschmidt-Clermont, P.J.; Finkel, T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 1996, 318, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Gorlach, A. NADPH oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Pothoulakis, C.; Castagliuolo, I.; Nikulasson, S.; LaMont, J.T. Participation of reactive oxygen metabolites in Clostridium difficile toxin A-induced enteritis in rats. Am. J. Physiol. 1999, 276, G485–G490. [Google Scholar] [PubMed]
- Farrow, M.A.; Chumbler, N.M.; Lapierre, L.A.; Franklin, J.L.; Rutherford, S.A.; Goldenring, J.R.; Lacy, D.B. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. USA 2013, 110, 18674–18679. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Sougioultzis, S.; Hagen, S.; Liu, J.; Keates, S.; Keates, A.C.; Pothoulakis, C.; Lamont, J.T. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 2002, 122, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Arbibe, L.; Mira, J.P.; Teusch, N.; Kline, L.; Guha, M.; Mackman, N.; Godowski, P.J.; Ulevitch, R.J.; Knaus, U.G. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol. 2000, 1, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Teusch, N.; Lombardo, E.; Eddleston, J.; Knaus, U.G. The low molecular weight GTPase RhoA and atypical protein kinase Czeta are required for TLR2-mediated gene transcription. J. Immunol. 2004, 173, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, L.Y.; Zuraw, B.L.; Ye, R.D.; Pan, Z.K. Chemoattractant-stimulated NF-kappaB activation is dependent on the low molecular weight GTPase RhoA. J. Biol. Chem. 2001, 276, 40977–40981. [Google Scholar] [CrossRef] [PubMed]
- Shen, A. Clostridium difficile toxins: Mediators of inflammation. J. Innate Immun. 2012, 4, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Hippenstiel, S.; Soeth, S.; Kellas, B.; Fuhrmann, O.; Seybold, J.; Krull, M.; Eichel-Streiber, C.; Goebeler, M.; Ludwig, S.; Suttorp, N. Rho proteins and the p38-MAPK pathway are important mediators for LPS-induced interleukin-8 expression in human endothelial cells. Blood 2000, 95, 3044–3051. [Google Scholar] [PubMed]
- Kim, H.; Kokkotou, E.; Na, X.; Rhee, S.H.; Moyer, M.P.; Pothoulakis, C.; Lamont, J.T. Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology 2005, 129, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Rhee, S.H.; Kokkotou, E.; Na, X.; Savidge, T.; Moyer, M.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J. Biol. Chem. 2005, 280, 21237–21245. [Google Scholar] [CrossRef] [PubMed]
- Linevsky, J.K.; Pothoulakis, C.; Keates, S.; Warny, M.; Keates, A.C.; Lamont, J.T.; Kelly, C.P. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am. Physiol. Soc. 1997, 273, G1333–G1340. [Google Scholar]
- Flegel, W.A.; Müller, F.; Däubener, W.; Fischer, H.G.; Hadding, U.; Northoff, H. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect. Immun. 1991, 59, 3659–3666. [Google Scholar] [PubMed]
- Balletta, A.; Lorenz, D.; Rummel, A.; Gerhard, R.; Bigalke, H.; Wegner, F. Clostridium difficile toxin B inhibits the secretory response of human mast cell ling-1 (HMC-1) cells stimulated with high free-Ca2+ and GTPγs. Toxicology 2014, 328, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Lyerly, D.M.; Lockwood, D.E.; Richardson, S.H.; Wilkins, T.D. Biological activities of toxins A and B of Clostridium difficile. Infect. Immun. 1982, 35, 1147–1150. [Google Scholar] [PubMed]
- Meyer, G.K.; Neetz, A.; Brandes, G.; Tsikas, D.; Butterfield, J.H.; Just, I.; Gerhard, R. Clostridium difficile toxins A and B directly stimulate human mast cells. Infect. Immun. 2007, 75, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- Riegler, M.; Sedivy, R.; Pothoulakis, C.; Hamilton, G.; Zacherl, J.; Bischof, G.; Cosentini, E.; Feil, W.; Schiessel, R.; LaMont, J.T.; et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in virto. J. Clin. Investig. 1995, 95, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Stabler, R.A.; Gerding, D.N.; Songer, J.G.; Drudy, D.; Brazier, J.S.; Trinh, H.T.; Witney, A.A.; Hinds, J.; Wren, B.W. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J. Bacteriol. 2006, 188, 7297–7305. [Google Scholar] [CrossRef] [PubMed]
- Alfa, M.J.; Kabani, A.; Lyerly, D.; Moncrief, S.; Neville, L.M.; Al-Barrak, A.; Harding, G.K.; Dyck, B.; Olekson, K.; Embil, J.M. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile responsible for a nosocomial outbreak of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 2000, 38, 2706–2714. [Google Scholar] [PubMed]
- Liu, M.; Bi, F.; Zhou, X.; Zheng, Y. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012, 22, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Visvikis, O.; Maddugoda, M.P.; Lemichez, E. Direct modifications of Rho proteins: Deconstructing GTPase regulation. Biol. Cell 2010, 102, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Garcia-Mata, R.; Burridge, K. Rho protein crosstalk: Another social network? Trends Cell Biol. 2011, 21, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Giry, M.; Popoff, M.R.; von Eichel-Streiber, C.; Boquet, P. Transient expression of RhoA, -B, and -C GTPases in HeLa cells potentiates resistance to Clostridium difficile toxins A and B but not to Clostridium sordellii lethal toxin. Infect. Immun. 1995, 63, 4063–4071. [Google Scholar] [PubMed]
- Santos, A.A.; Braga-Neto, M.B.; Oliveira, M.R.; Freire, R.S.; Barros, E.B.; Santiago, T.M.; Rebelo, L.M.; Mermelstein, C.; Warren, C.A.; Guerrant, R.L.; et al. Glutamine and alanyl-glutamine increase RhoA expression and reduce Clostridium difficile toxin-a-induced intestinal epithelial cell damage. BioMed Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Richter, H.P.; Prepens, U.; von Eichel-Streiber, C.; Aktories, K. Probing the action of Clostridium difficile toxin B in Xenopus laevis oocytes. J. Cell Sci. 1994, 107, 1653–1659. [Google Scholar] [PubMed]
- Brandes, V.; Schelle, I.; Brinkmann, S.; Schulz, F.; Schwarz, J.; Gerhard, R.; Genth, H. Protection from Clostridium difficile toxin B-catalysed Rac1/Cdc42 glucosylation by tauroursodeoxycholic acid-induced Rac1/Cdc42 phosphorylation. Biol. Chem. 2012, 393, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Schoentaube, J.; Olling, A.; Tatge, H.; Just, I.; Gerhard, R. Serine-71 phosphorylation of Rac1/Cdc42 diminishes the pathogenic effect of Clostridium difficile toxin A. Cell. Microbiol. 2009, 11, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Viladomiu, M.; Hontecillas, R.; Pedragosa, M.; Carbo, A.; Hoops, S.; Michalak, P.; Michalak, K.; Guerrant, R.L.; Roche, J.K.; Warren, C.A.; et al. Modeling the role of peroxisome proliferator-activated receptor gamma and microRNA-146 in mucosal immune responses to Clostridium difficile. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Mraheil, M.A.; Billion, A.; Kuenne, C.; Pischimarov, J.; Kreikemeyer, B.; Engelmann, S.; Hartke, A.; Giard, J.C.; Rupnik, M.; Vorwerk, S.; et al. Comparative genome-wide analysis of small RNAs of major Gram-positive pathogens: From identification to application. Microb. Biotechnol. 2010, 3, 658–676. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Sun, C.; Wang, H.; Wang, J. The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins. Toxins 2015, 7, 5254-5267. https://doi.org/10.3390/toxins7124874
Chen S, Sun C, Wang H, Wang J. The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins. Toxins. 2015; 7(12):5254-5267. https://doi.org/10.3390/toxins7124874
Chicago/Turabian StyleChen, Shuyi, Chunli Sun, Haiying Wang, and Jufang Wang. 2015. "The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins" Toxins 7, no. 12: 5254-5267. https://doi.org/10.3390/toxins7124874
APA StyleChen, S., Sun, C., Wang, H., & Wang, J. (2015). The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins. Toxins, 7(12), 5254-5267. https://doi.org/10.3390/toxins7124874