
A Simple and Rapid Procedure for the Detection of Genes Encoding Shiga Toxins and Other Specific DNA Sequences




Abstract
:
1. Introduction
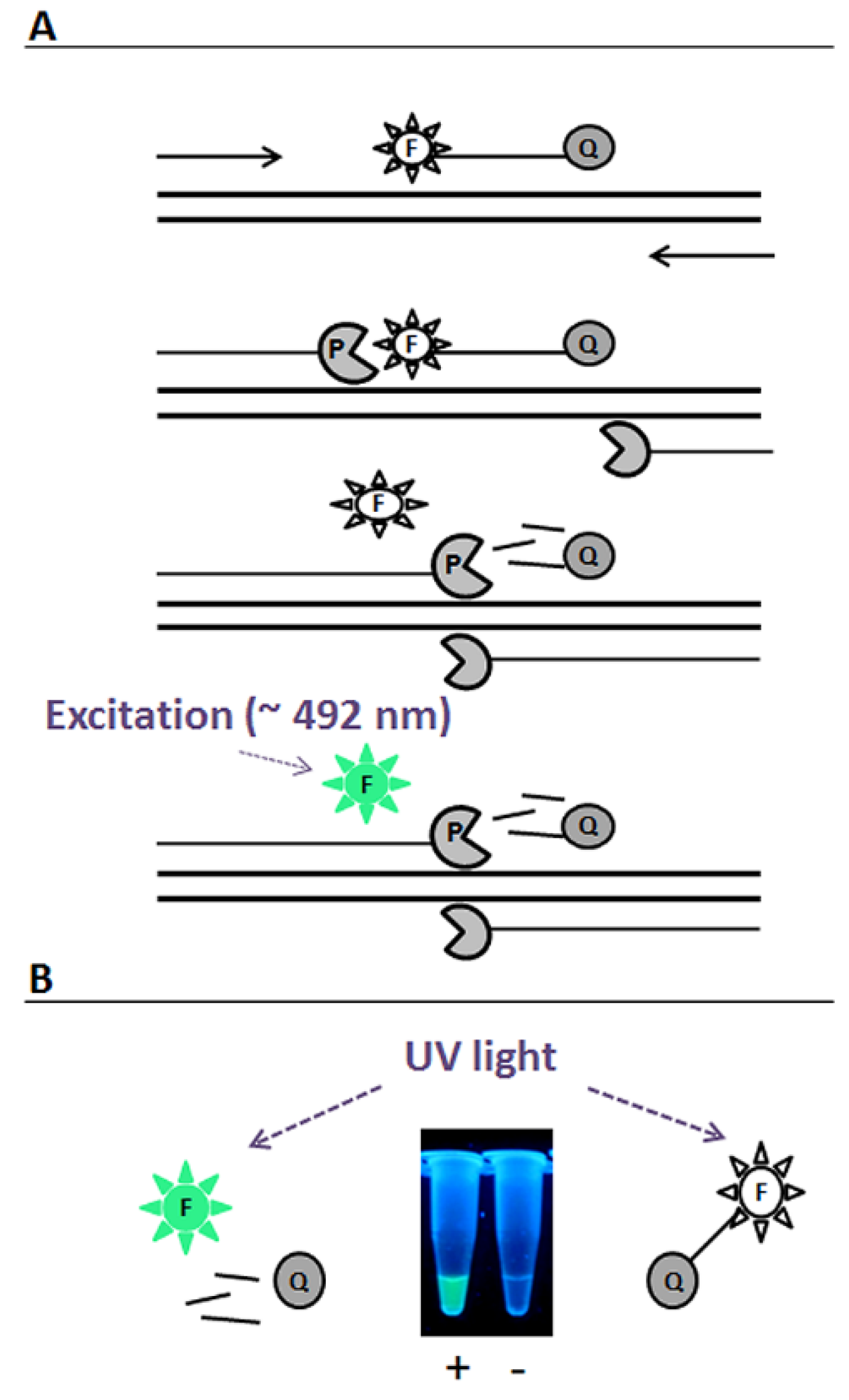
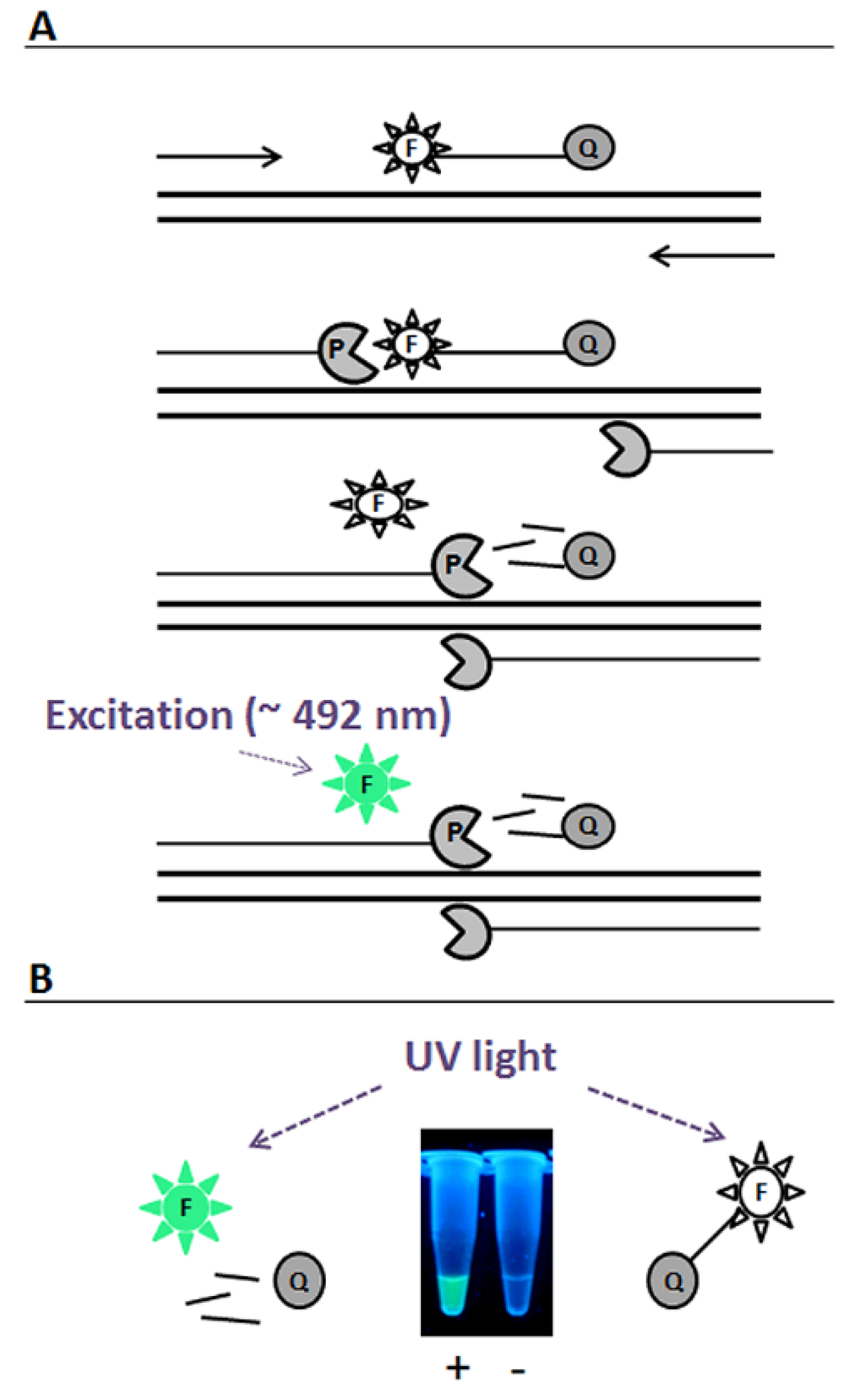
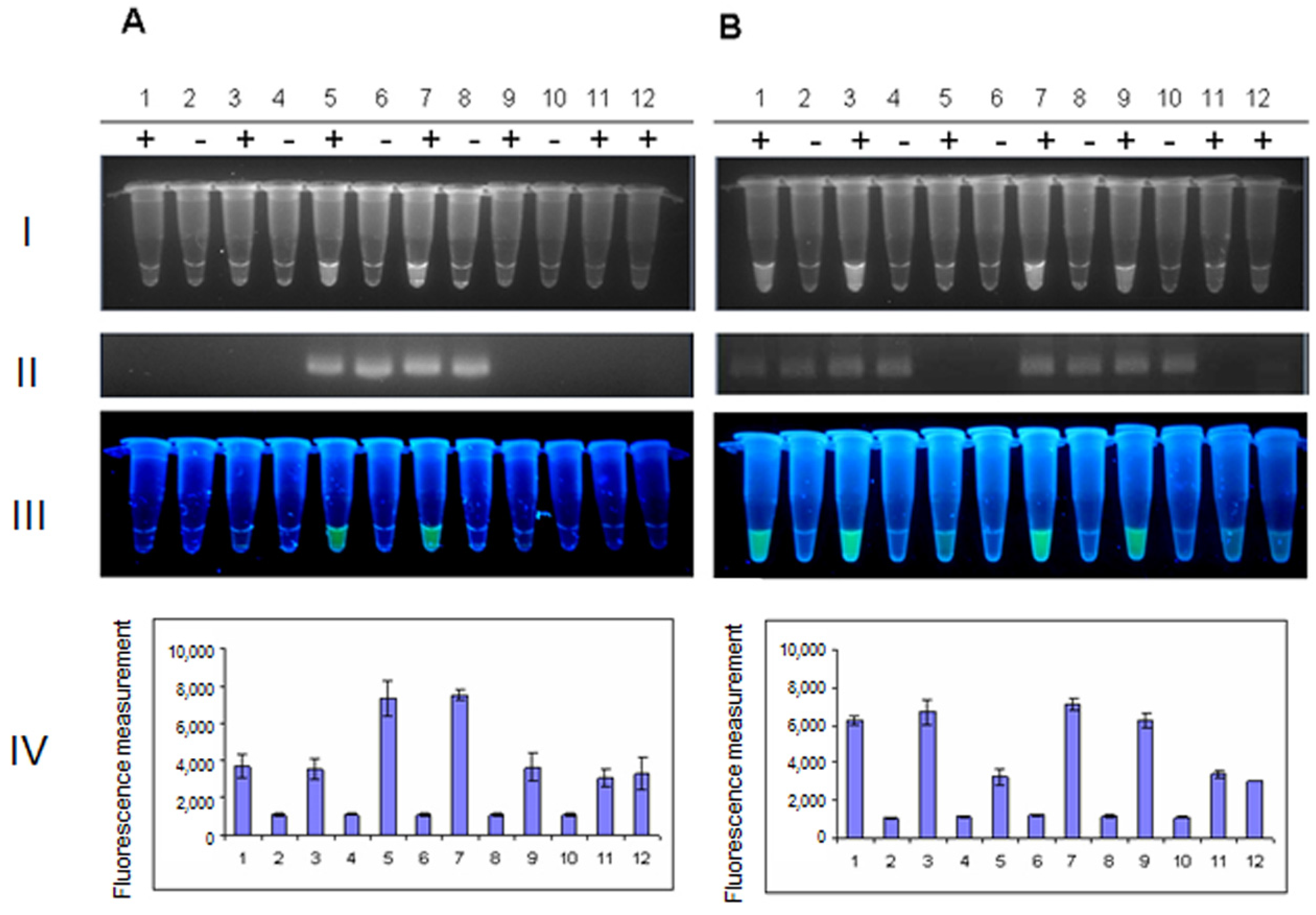
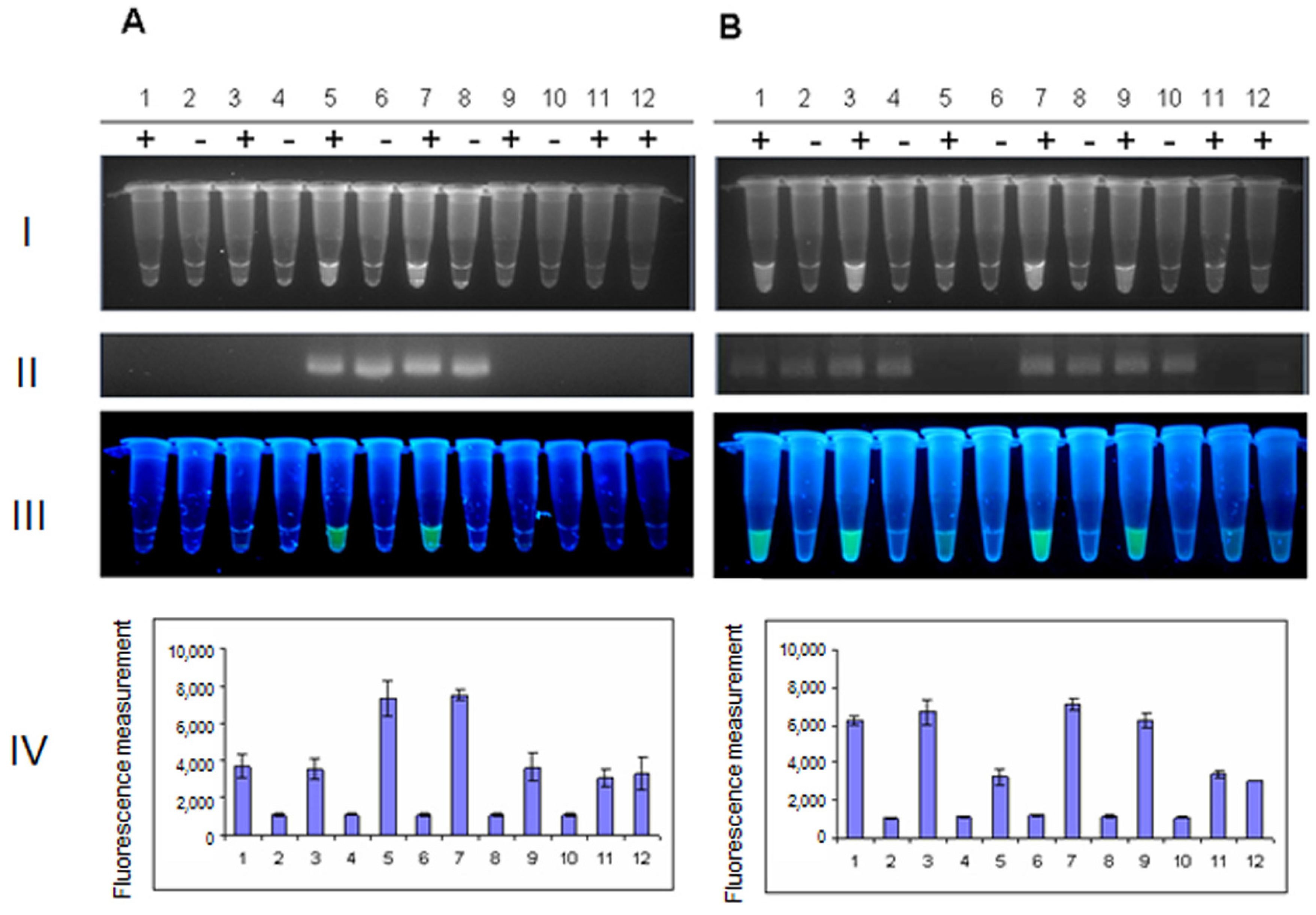
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
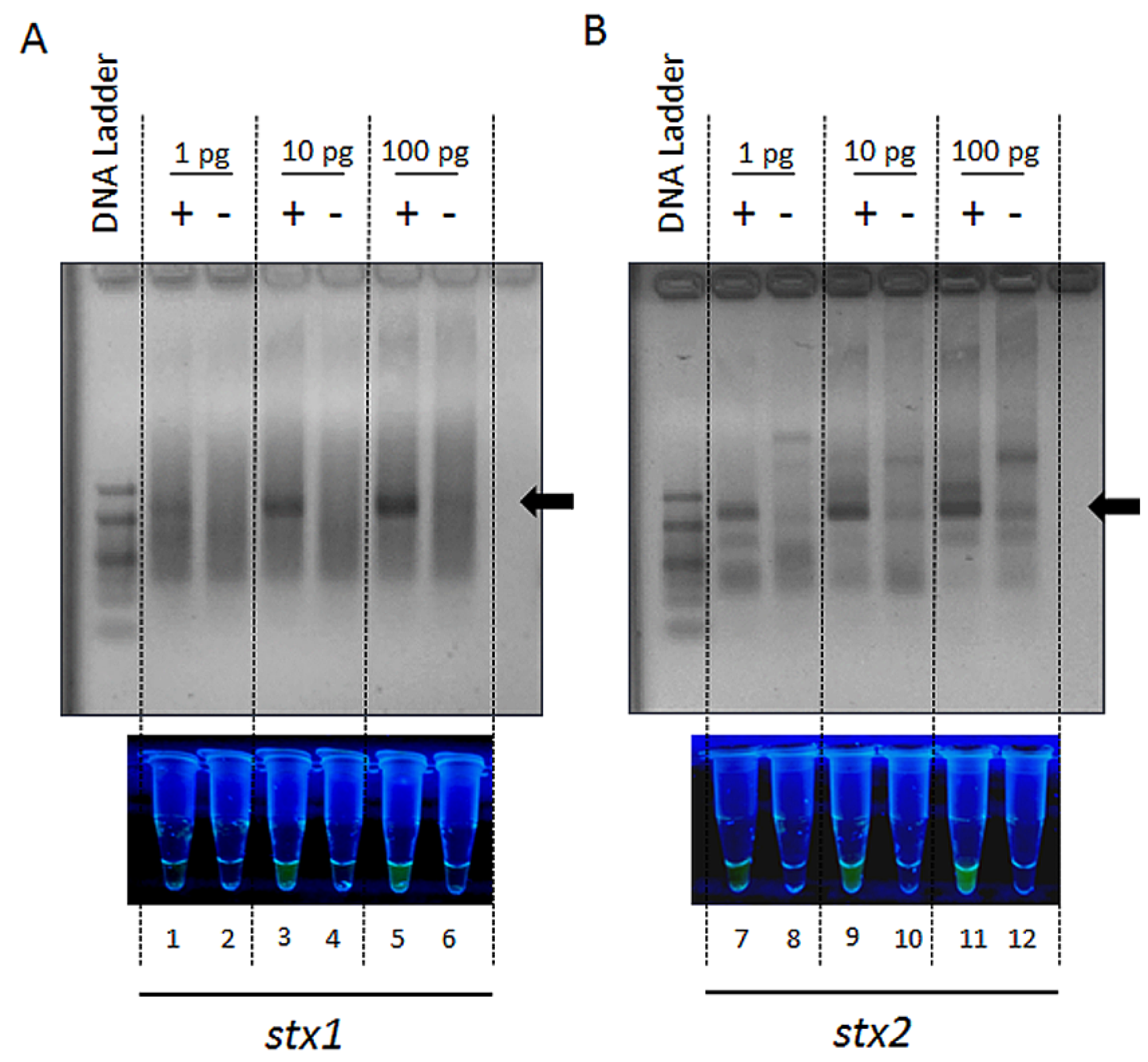{kind=link}
{kind=link}
| No. | Strain | Serotype | Stx1 Status | Stx2 Status |
|---|---|---|---|---|
| 1 | 286/00 | O157 | − | + |
| 2 | 44/02 | O157 | + | + |
| 3 | 174/03 | O157 | − | + |
| 4 | 49/04 | O157 | + | + |
| 5 | 365/05 | O157 | + | + |
| 6 | 206/06 | O157 | + | + |
| 7 | 443/07 | O157 | + | + |
| 8 | 474/07 | O157 | + | + |
| 9 | 9/08 | O157 | − | + |
| 10 | 221/08 | O157 | + | + |
| 11 | 371/08 | O157 | − | + |
| 12 | 171/09 | O157 | − | + |
| 13 | 74/10 | O157 | − | + |
| 14 | 245/10 | O111 | − | + |
| 15 | 251/10 | O157 | + | + |
| 16 | 201/01 | O26 | − | + |
| 17 | 319/01 | O26 | + | − |
| 18 | 571 | O157 | + | + |
| 19 | EDL933W | O157 | + | + |
| 20 | MG1655 | K12 | − | − |
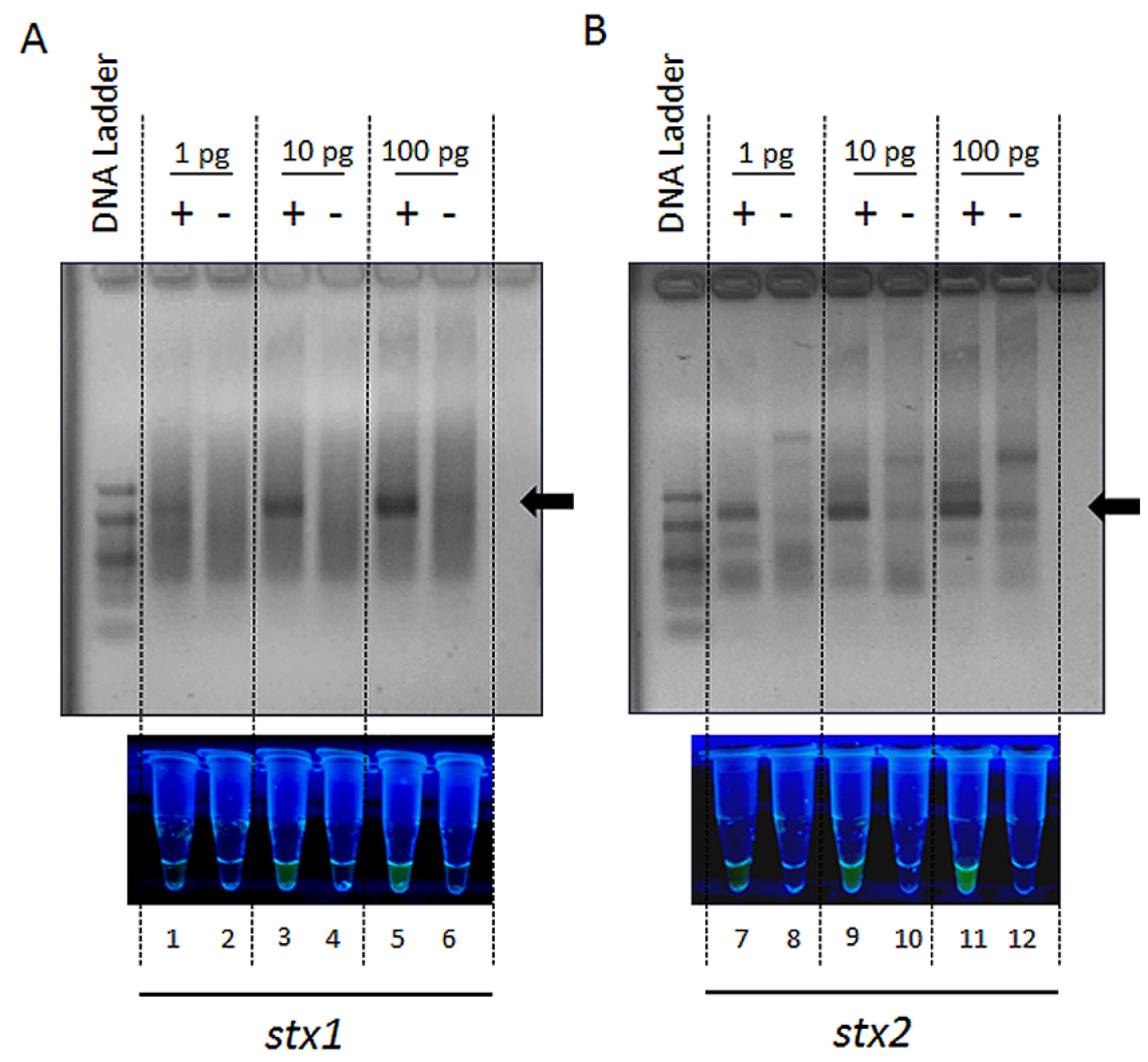
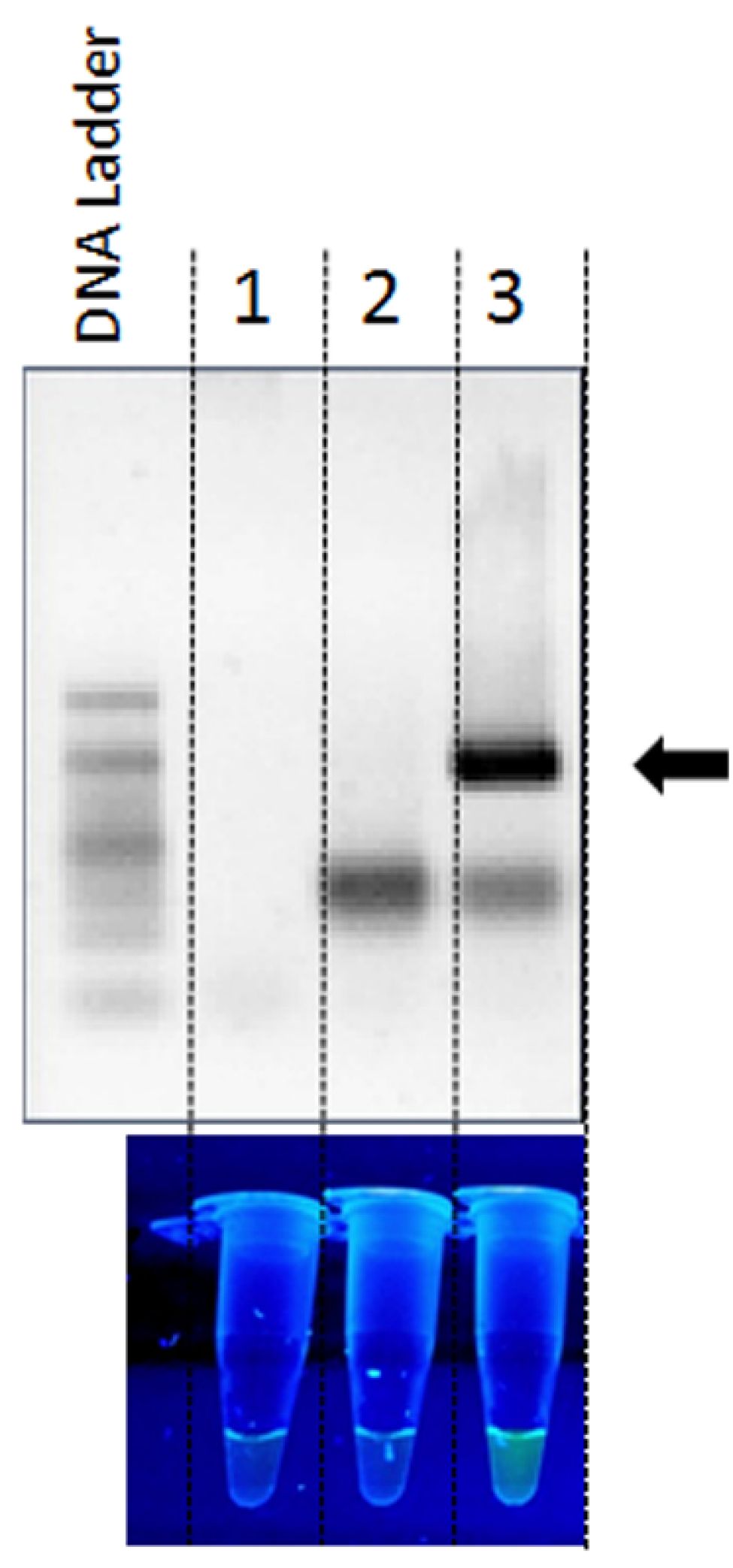
3. Experimental Section
3.1. Bacterial Strains
3.2. DNA Amplification and Detection of Signals Specific for Particular DNA Sequences
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Armua-Fernandez, M.T.; Nonaka, N.; Sakurai, T.; Nakamura, S.; Gottstein, B.; Deplazes, P.; Phiri, I.G.; Katakura, K.; Oku, Y. Development of PCR/dot blot assay for specific detection and differentiation of taeniid cestode eggs in canids. Parasitol. Int. 2011, 60, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Curry, E.; Pratt, S.L.; Kelley, D.E.; Lapin, D.R.; Gibbons, J.R. Use of a Combined Duplex PCR/Dot Blot Assay for more sensitive genetic characterization. Biochem. Insights 2008, 1, 35–39. [Google Scholar]
- Boerner, B.; Weigelt, W.; Buhk, H.J.; Castrucci, G.; Ludwig, H. A sensitive and specific PCR/Southern blot assay for detection of bovine herpesvirus 4 in calves infected experimentally. J. Virol. Methods 1999, 83, 169–180. [Google Scholar] [CrossRef]
- Ryschkewitsch, C.; Jensen, P.; Hou, J.; Fahle, G.; Fischer, S.; Major, E.O. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J. Virol. Methods 2004, 121, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Yeh, T.J.; Chang, Y.C. A new combination of RT-PCR and reverse dot blot hybridization for rapid detection and identification of potyviruses. J. Virol. Methods 2005, 128, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Gilbert, G.L. Multiplex PCR-based reverse line blot hybridization assay (mPCR/RLB)—a practical epidemiological and diagnostic tool. Nat. Protoc. 2007, 1, 2668–2680. [Google Scholar] [CrossRef] [PubMed]
- Jantos, C.A.; Roggendorf, R.; Wuppermann, F.N.; Hegemann, J.H. Rapid detection of Chlamydia pneumoniae by PCR-enzyme immunoassay. J. Clin. Microbiol. 1998, 36, 1890–1894. [Google Scholar] [PubMed]
- Mantero, G.; Zonaro, A.; Albertini, A.; Bertolo, P.; Primi, D. DNA enzyme immunoassay: General method for detecting products of polymerase chain reaction. Clin. Chem. 1991, 37, 422–429. [Google Scholar] [PubMed]
- Osawa, Y.; Ikebukuro, K.; Motoki, H.; Matsuo, T.; Horiuchi, M.; Sode, K. The simple and rapid detection of specific PCR products from bacterial genomes using Zn finger proteins. Nucleic Acids Res. 2008, 36, e68. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.M.S.; Stirling, D. Methods in Molecular Biology: PCR Protocols, 2nd ed.; Humana Press: New York, NY, USA, 2003. [Google Scholar]
- Findlay, J.B.; Atwood, S.M.; Bergmeyer, L.; Chemelli, J.; Christy, K.; Cummins, T.; Donish, W.; Ekeze, T.; Falvo, J.; Patterson, D. Automated closed-vessel system for in vitro diagnostics based on polymerase chain reaction. Clin. Chem. 1993, 39, 1927–1933. [Google Scholar] [PubMed]
- Kai, E.; Ikebukuro, K.; Hoshina, S.; Watanabe, H.; Karube, I. Detection of PCR products of Escherichia coli O157:H7 in human stool samples using surface plasmon resonance (SPR). FEMS Immunol. Med. Microbiol. 2000, 29, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.M.; Law, S.M.; Duan, S.; Neri, B.P.; Kwok, P.Y. Genotyping single-nucleotide polymorphisms by the invader assay with dual-color fluorescence polarization detection. Clin. Chem. 2001, 47, 1373–1377. [Google Scholar] [PubMed]
- Espy, M.J.; Uhl, J.R.; Sloan, L.M.; Buckwalter, S.P.; Jones, M.F.; Vetter, E.A.; Yao, J.D.; Wengenack, N.L.; Rosenblatt, J.E.; Cockerill, F.R.; et al. Real-time PCR in clinical microbiology: Applications for routine laboratory testing. Clin. Microbiol. Rev. 2006, 19, 165–256. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Zhang, Y.; Lin, S.; Wang, T.H.; Yang, S. Advances in microfluidic PCR for point-of-care infectious disease diagnostics. Biotechnol. Adv. 2011, 29, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Flood, S.J.; Marmaro, J.; Giusti, W.; Deetz, K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995, 4, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.C.; Davis, C.R.; Peak, K.K.; Wingfield, D.; Cannons, A.C.; Amuso, P.T.; Cattani, J. Comparison of methods for DNA isolation from food samples for detection of Shiga toxin-producing Escherichia coli by real-time PCR. Appl. Environ. Microbiol. 2003, 69, 1844–1846. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Didenko, V.V. DNA probes using fluorescence resonance energy transfer (FRET): Designs and applications. Biotechniques 2001, 31, 1106–1121. [Google Scholar] [PubMed]
- Gelmini, S.; Orlando, C.; Sestini, R.; Vona, G.; Pinzani, P.; Ruocco, L.; Pazzagli, M. Quantitative polymerase chain reaction-based homogeneous assay with fluorogenic probes to measure c-erbB-2 oncogene amplification. Clin. Chem. 1997, 43, 752–758. [Google Scholar] [PubMed]
- Latif, S.; Bauer-Sardina, I.; Ranade, K.; Livak, K.J.; Kwok, P.Y. Fluorescence polarization in homogeneous nucleic acid analysis II: 5′-nuclease assay. Genome Res. 2001, 11, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.M. Shiga toxin-producing Escherichia coli (STEC). Clin. Lab. Med. 2010, 30, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Bloch, S.K.; Felczykowska, A.; Nejman-Faleńczyk, B. Escherichia coli O104:H4 outbreak-have we learnt a lesson from it? Acta. Biochim. Pol. 2012, 59, 483–488. [Google Scholar] [PubMed]
- Gyles, C.L. Shiga toxin-producing Escherichia coli: An overview. J. Anim. Sci. 2007, 85, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Serna, A.; Boedeker, E.C. Pathogenesis and treatment of Shiga toxin-producing Escherichia coli infections. Curr. Opin. Gastroenterol. 2008, 24, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Harmsen, D.; Cummings, C.A.; Zentz, E.B.; Leopold, S.R.; Rico, A.; Prior, K.; Szczepanowski, R.; Ji, Y.; Zhang, W.; et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE 2011, 6, e22751. [Google Scholar] [CrossRef] [PubMed]
- Beutin, L.; Martin, A. Outbreak of Shiga toxin-producing Escherichia coli (STEC) O104:H4 infection in Germany causes a paradigm shift with regard to human pathogenicity of STEC strains. J. Food Prot. 2012, 75, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Karch, H.; Denamur, E.; Dobrindt, U.; Finlay, B.B.; Hengge, R.; Johannes, L.; Ron, E.Z.; Tønjum, T.; Sansonetti, P.J.; Vicente, M. The enemy within us: Lessons from the 2011 European Escherichia coli O104:H4 outbreak. EMBO Mol. Med. 2012, 4, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Werber, D.; Krause, G.; Frank, C.; Fruth, A.; Flieger, A.; Mielke, M.; Schaade, L.; Stark, K. Outbreaks of virulent diarrheagenic Escherichia coli-are we in control? BMC Med. 2012, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Łoś, J.M.; Łoś, M.; Węgrzyn, G. Bacteriophages carrying Shiga toxin genes: Genomic variations, detection and potential treatment of pathogenic bacteria. Future Microbiol. 2011, 6, 909–924. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.J.; Jin, D.Z.; Luo, Y.; Li, H.; Zheng, Y.; Zhang, Z.; Gu, H.; Zheng, S.S. A DNA minor groove binder shows high effectiveness as a quencher for FRET probes. Bioorg. Med. Chem. Lett. 2014, 24, 3956–3960. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; James, C.E.; Sergeant, M.J.; Yaxian, Y.; Saunders, J.R.; McCarthy, A.J.; Allison, H.E. Short-tailed stx phages exploit the conserved YaeT protein to disseminate Shiga toxin genes among enterobacteria. J. Bacteriol. 2007, 189, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.F.; Du, M.; Zhu, M.J. High temperature in combination with UV irradiation enhances horizontal transfer of stx2 gene from E. coli O157:H7 to non-pathogenic E. coli. PLoS ONE 2012, 7, e31308. [Google Scholar] [PubMed]
- Fagan, P.K.; Hornitzky, M.A.; Bettelheim, K.A.; Djordjevic, S.P. Detection of shiga-like toxin (stx1 and stx2), intimin (eaeA), and enterohemorrhagic Escherichia coli (EHEC) hemolysin (EHEC hlyA) genes in animal feces by multiplex PCR. Appl. Environ. Microbiol. 1999, 65, 868–872. [Google Scholar] [PubMed]
- Botkin, D.J.; Galli, L.; Sankarapani, V.; Soler, M.; Rivas, M.; Torres, A.G. Development of a multiplex PCR assay for detection of Shiga toxin-producing Escherichia coli, enterohemorrhagic E. coli, and enteropathogenic E. coli strains. Front. Cell. Infect. Microbiol. 2012, 14, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, A.J.; Steeves, R.; Newmaster, S.G. Improving sequencing quality from PCR products containing long mononucleotide repeats. BioTechniques 2010, 48, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, T.C. Polymerase Chain Reaction: Basic Protocol Plus Troubleshooting and Optimization Strategies. J. Vis. Exp. 2012, 63, e3998. [Google Scholar] [CrossRef] [PubMed]
- Massung, R.F.; Slater, K.G. Comparison of PCR assays for detection of the agent of human granulocytic ehrlichiosis, Anaplasma. phagocytophilum. J. Clin. Microbiol. 2003, 41, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Qurollo, B.A.; Riggins, D.; Comyn, A.; Zewde, M.T.; Breitschwerdt, E.B. Development and validation of a sensitive and specific sodB-based quantitative PCR assay for molecular detection of Ehrlichia. species. J. Clin. Microbiol. 2014, 52, 4030–4032. [Google Scholar] [CrossRef] [PubMed]
- Konowalchuk, J.; Speirs, J.I.; Stavric, S. Vero response to a cytotoxin of Escherichia coli. Infect. Immun. 1977, 18, 775–779. [Google Scholar] [PubMed]
- Januszkiewicz, A.; Wołkowicz, T.; Chróst, A.; Szych, J. Characterization of the Shiga toxin-producing Escherichia coli O26 isolated from human in Poland between 1996 and 2014. Lett. Appl. Microbiol. 2015, 60, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Rüssmann, H.; Kothe, E.; Schmidt, H.; Franke, S.; Harmsen, D.; Caprioli, A.; Karch, H. Genotyping of Shiga-like toxin genes in non-O157 Escherichia coli strains associated with haemolytic uraemic syndrome. J. Med. Microbiol. 1995, 42, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Januszkiewicz, A.; Szych, J.; Rastawicki, W.; Wołkowicz, T.; Chróst, A.; Leszczyńska, B.; Kuźma, E.; Roszkowska-Blaim, M.; Gierczyński, R. Molecular epidemiology of a household outbreak of Shiga-toxin-producing Escherichia coli in Poland due to secondary transmission of STEC O104:H4 from Germany. J. Med. Microbiol. 2012, 61, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Queipo-Ortuño, M.I.; De Dios Colmenero, J.; Macias, M.; Bravo, M.J.; Morata, P. Preparation of bacterial DNA template by boiling and effect of immunoglobulin G as an inhibitor in real-time PCR for serum samples from patients with brucellosis. Clin. Vaccine Immunol. 2008, 15, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Gierczyński, R.; Kałuzewski, S.; Rakin, A.; Jagielski, M.; Zasada, A.; Jakubczak, A.; Borkowska-Opacka, B.; Rastawicki, W. Intriguing diversity of Bacillus anthracis in eastern Poland-the molecular echoes of the past outbreaks. FEMS Microbiol. Lett. 2004, 239, 235–240. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nejman-Faleńczyk, B.; Bloch, S.; Januszkiewicz, A.; Węgrzyn, A.; Węgrzyn, G. A Simple and Rapid Procedure for the Detection of Genes Encoding Shiga Toxins and Other Specific DNA Sequences. Toxins 2015, 7, 4745-4757. https://doi.org/10.3390/toxins7114745
Nejman-Faleńczyk B, Bloch S, Januszkiewicz A, Węgrzyn A, Węgrzyn G. A Simple and Rapid Procedure for the Detection of Genes Encoding Shiga Toxins and Other Specific DNA Sequences. Toxins. 2015; 7(11):4745-4757. https://doi.org/10.3390/toxins7114745
Chicago/Turabian StyleNejman-Faleńczyk, Bożena, Sylwia Bloch, Aleksandra Januszkiewicz, Alicja Węgrzyn, and Grzegorz Węgrzyn. 2015. "A Simple and Rapid Procedure for the Detection of Genes Encoding Shiga Toxins and Other Specific DNA Sequences" Toxins 7, no. 11: 4745-4757. https://doi.org/10.3390/toxins7114745
APA StyleNejman-Faleńczyk, B., Bloch, S., Januszkiewicz, A., Węgrzyn, A., & Węgrzyn, G. (2015). A Simple and Rapid Procedure for the Detection of Genes Encoding Shiga Toxins and Other Specific DNA Sequences. Toxins, 7(11), 4745-4757. https://doi.org/10.3390/toxins7114745