Immunotoxins: The Role of the Toxin †


{kind=link}
{kind=link}
Abstract
:1. Introduction
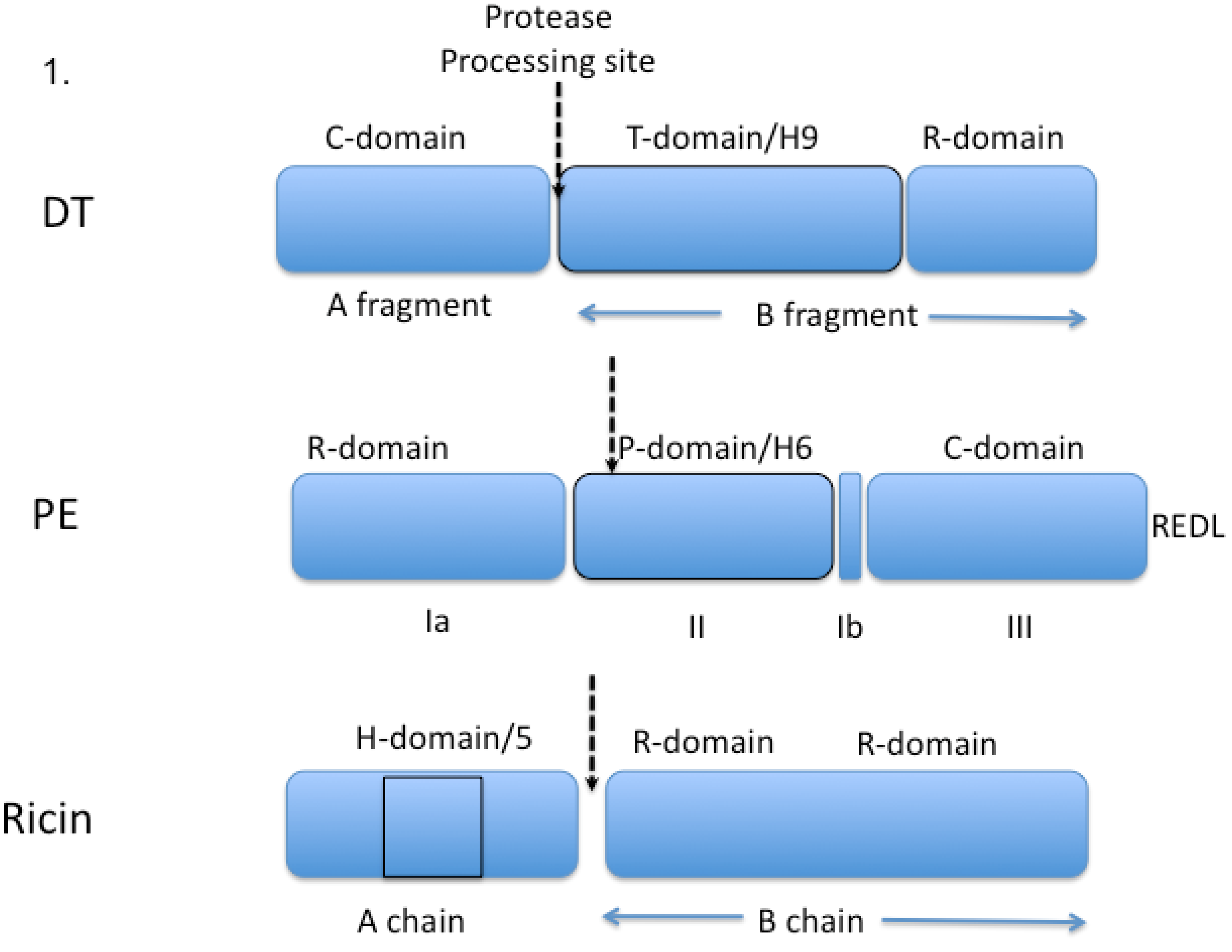
2. Toxin Candidates
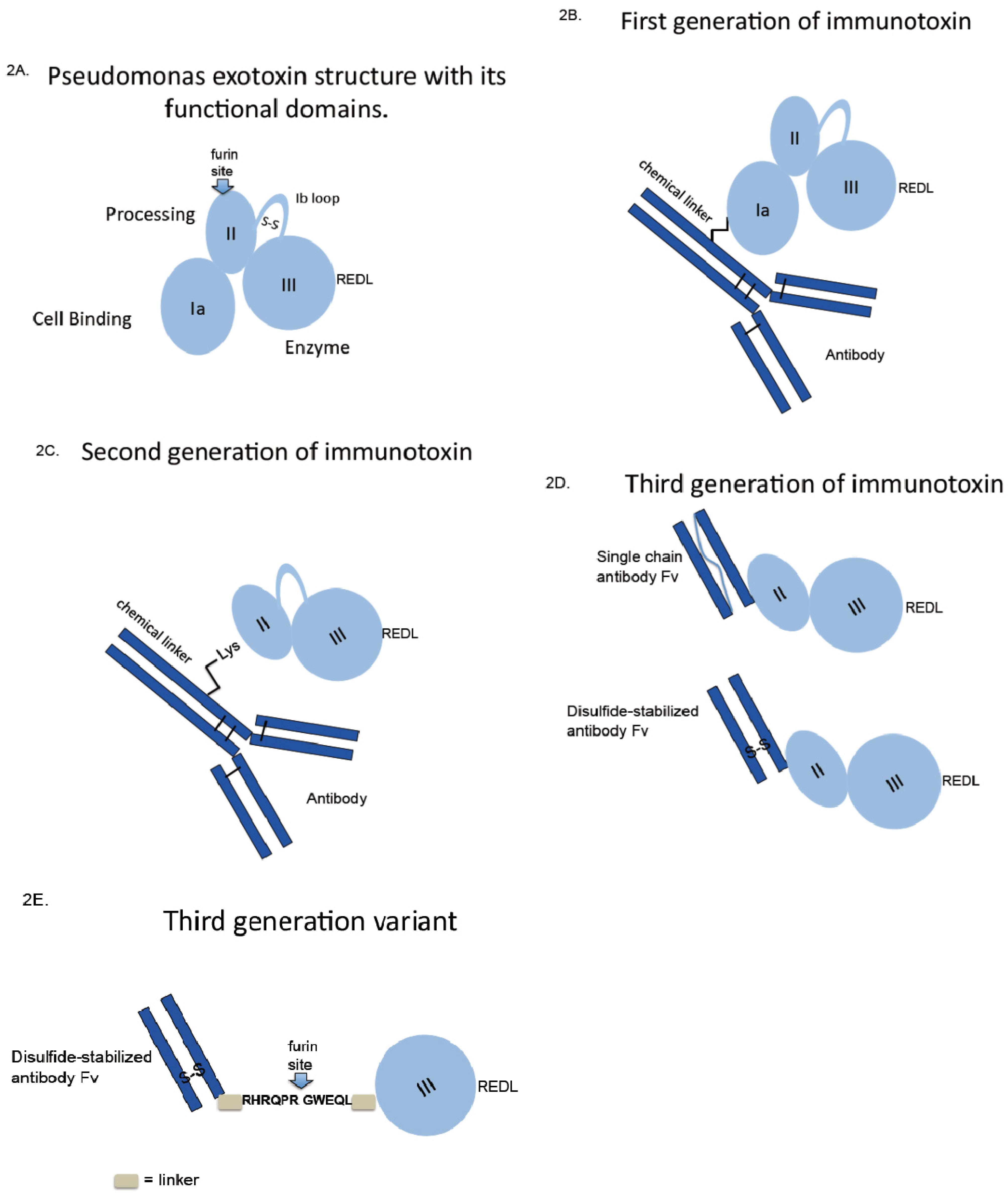
3. Early Immunotoxin Development
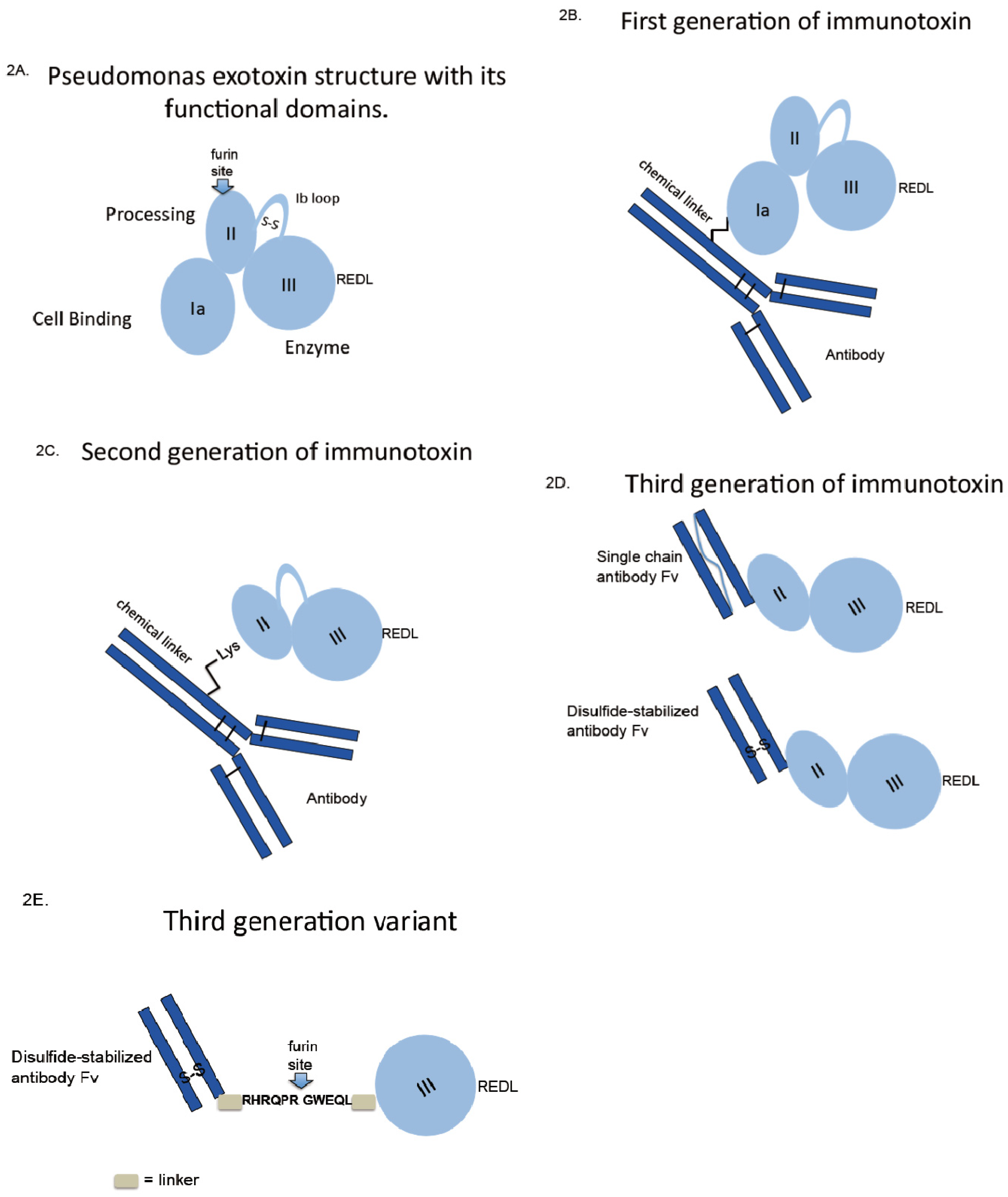
4. Evolved Immunotoxins
5. Gene Fusions with Cell-Binding Ligands
6. Toxin Resistance
7. Immunotoxin-Drug Combinations
8. Future Directions
9. Conclusions
Acknowledgements
Conflicts of Interest
References
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Thorpe, P.E.; Ross, W.C.; Cumber, A.J.; Hinson, C.A.; Edwards, D.C.; Davies, A.J. Toxicity of diphtheria toxin for lymphoblastoid cells is increased by conjugation to antilymphocytic globulin. Nature 1978, 271, 752–755. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Krolick, K.A.; Miyama-Inaba, M.; Cushley, W.; Uhr, J.W. Immunotoxins: A new approach to cancer therapy. Science 1983, 219, 644–650. [Google Scholar]
- Vitetta, E.S.; Krolick, K.A.; Uhr, J.W. Neoplastic B cells as targets for antibody-ricin A chain immunotoxins. Immunol. Rev. 1982, 62, 159–183. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Uhr, J.W. Immunotoxins: Redirecting nature’s poisons. Cell 1985, 41, 653–654. [Google Scholar] [CrossRef]
- Pastan, I.; Willingham, M.C.; FitzGerald, D.J. Immunotoxins. Cell 1986, 47, 641–648. [Google Scholar] [CrossRef]
- Neville, D.M., Jr.; Youle, R.J. Monoclonal antibody-ricin or ricin A chain hybrids: Kinetic analysis of cell killing for tumor therapy. Immunol. Rev. 1982, 62, 75–91. [Google Scholar] [CrossRef]
- FitzGerald, D.; Pastan, I. Redirecting Pseudomonas exotoxin. Semin. Cell Biol. 1991, 2, 31–37. [Google Scholar]
- Pastan, I.; FitzGerald, D. Recombinant toxins for cancer treatment. Science 1991, 254, 1173–1177. [Google Scholar]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; DeVroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1051. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro-Oncology 2010, 12, 871–881. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.M.; Prados, M.D.; Berger, M.S.; Sampson, J.H.; Croteau, D.; Sherman, J.W.; Grahn, A.Y.; Shu, V.S.; Dul, J.L.; et al. Safety of intraparenchymal convection-enhanced delivery of cintredekin besudotox in early-phase studies. Neurosurg. Focus 2006, 20, 15. [Google Scholar]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; Wilson, W.H.; Pastan, I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutre, S.; Wilson, W.H.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Lechleider, R.; Pastan, I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol. 2012, 30, 1822–1828. [Google Scholar] [CrossRef]
- Wayne, A.S.; Kreitman, R.J.; Findley, H.W.; Lew, G.; Delbrook, C.; Steinberg, S.M.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Pastan, I. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: Preclinical studies and phase I clinical trial. Clin. Cancer Res. 2010, 16, 1894–1903. [Google Scholar] [CrossRef]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin. Cancer Res. 2000, 6, 1302–1313. [Google Scholar]
- Herrera, L.; Bostrom, B.; Gore, L.; Sandler, E.; Lew, G.; Schlegel, P.G.; Aquino, V.; Ghetie, V.; Vitetta, E.S.; Schindler, J. A phase 1 study of Combotox in pediatric patients with refractory B-lineage acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2009, 31, 936–941. [Google Scholar] [CrossRef]
- Schindler, J.; Gajavelli, S.; Ravandi, F.; Shen, Y.; Parekh, S.; Braunchweig, I.; Barta, S.; Ghetie, V.; Vitetta, E.; Verma, A. A phase I study of a combination of anti-CD19 and anti-CD22 immunotoxins (Combotox) in adult patients with refractory B-lineage acute lymphoblastic leukaemia. Br. J. Haematol. 2011, 154, 471–476. [Google Scholar] [CrossRef]
- Grossbard, M.L.; Fidias, P.; Kinsella, J.; O’Toole, J.; Lambert, J.M.; Blattler, W.A.; Esseltine, D.; Braman, G.; Nadler, L.M.; Anderson, K.C. Anti-B4-blocked ricin: A phase II trial of 7 day continuous infusion in patients with multiple myeloma. Br. J. Haematol. 1998, 102, 509–515. [Google Scholar] [CrossRef]
- Multani, P.S.; O’Day, S.; Nadler, L.M.; Grossbard, M.L. Phase II clinical trial of bolus infusion anti-B4 blocked ricin immunoconjugate in patients with relapsed B-cell non-Hodgkin’s lymphoma. Clin. Cancer Res. 1998, 4, 2599–2604. [Google Scholar]
- Furman, R.R.; Grossbard, M.L.; Johnson, J.L.; Pecora, A.L.; Cassileth, P.A.; Jung, S.H.; Peterson, B.A.; Nadler, L.M.; Freedman, A.; Bayer, R.L.; et al. A phase III study of anti-B4-blocked ricin as adjuvant therapy post-autologous bone marrow transplant: CALGB 9254. Leuk. Lymphoma. 2011, 52, 587–596. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N.Engl.J.Med. 2001, 345, 241–247. [Google Scholar] [CrossRef]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin.Cancer Res. 2007, 13, 5144–5149. [Google Scholar]
- Foss, F.M.; Saleh, M.N.; Krueger, J.G.; Nichols, J.C.; Murphy, J.R. Diphtheria toxin fusion proteins. Curr. Top. Microbiol. Immunol. 1998, 234, 63–81. [Google Scholar] [CrossRef]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar]
- Casellas, P.; Canat, X.; Fauser, A.A.; Gros, O.; Laurent, G.; Poncelet, P.; Jansen, F.K. Optimal elimination of leukemic T cells from human bone marrow with T101-ricin A-chain immunotoxin. Blood 1985, 65, 289–297. [Google Scholar]
- Fulton, R.J.; Uhr, J.W.; Vitetta, E.S. The effect of antibody valency and lysosomotropic amines on the synergy between ricin A chain- and ricin B chain-containing immunotoxins. J. Immunol. 1986, 136, 3103–3109. [Google Scholar]
- Hudson, T.H.; Neville, D.M., Jr. Enhancement of immunotoxin action: Manipulation of the cellular routing of proteins. Cancer Treat. Res. 1988, 37, 371–389. [Google Scholar]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef]
- Stong, R.C.; Uckun, F.; Youle, R.J.; Kersey, J.H.; Vallera, D.A. Use of multiple T cell-directed intact ricin immunotoxins for autologous bone marrow transplantation. Blood 1985, 66, 627–635. [Google Scholar]
- Vallera, D.A.; Burns, L.J.; Frankel, A.E.; Sicheneder, A.R.; Gunther, R.; Gajl-Peczalska, K.; Pennell, C.A.; Kersey, J.H. Laboratory preparation of a deglycosylated ricin toxin A chain containing immunotoxin directed against a CD7 T lineage differentiation antigen for phase I human clinical studies involving T cell malignancies. J. Immunol. Methods 1996, 197, 69–83. [Google Scholar] [CrossRef]
- Thompson, J.; Hu, H.; Scharff, J.; Neville, D.M., Jr. An anti-CD3 single-chain immunotoxin with a truncated diphtheria toxin avoids inhibition by pre-existing antibodies in human blood. J. Biol. Chem. 1995, 270, 28037–28041. [Google Scholar]
- Weetall, M.; Digan, M.E.; Hugo, R.; Mathew, S.; Hopf, C.; Tart-Risher, N.; Zhang, J.; Shi, V.; Fu, F.; Hammond-McKibben, D.; et al. T-cell depletion and graft survival induced by anti-human CD3 immunotoxins in human CD3epsilon transgenic mice. Transplantation 2002, 73, 1658–1666. [Google Scholar] [CrossRef]
- Martin, P.J.; Nelson, B.J.; Appelbaum, F.R.; Anasetti, C.; Deeg, H.J.; Hansen, J.A.; McDonald, G.B.; Nash, R.A.; Sullivan, K.M.; Witherspoon, R.P.; et al. Evaluation of a CD5-specific immunotoxin for treatment of acute graft-versus-host disease after allogeneic marrow transplantation. Blood 1996, 88, 824–830. [Google Scholar]
- Martin, P.J.; Pei, J.; Gooley, T.; Anasetti, C.; Appelbaum, F.R.; Deeg, J.; Hansen, J.A.; Nash, R.A.; Petersdorf, E.W.; Storb, R.; et al. Evaluation of a CD25-specific immunotoxin for prevention of graft-versus-host disease after unrelated marrow transplantation. Biol. Blood Marrow. Transplant. 2004, 10, 552–560. [Google Scholar] [CrossRef]
- Powell, D.J., Jr.; Attia, P.; Ghetie, V.; Schindler, J.; Vitetta, E.S.; Rosenberg, S.A. Partial reduction of human FOXP3+ CD4 T cells in vivo after CD25-directed recombinant immunotoxin administration. J. Immunother. 2008, 31, 189–198. [Google Scholar] [CrossRef]
- Powell, D.J., Jr.; Felipe-Silva, A.; Merino, M.J.; Ahmadzadeh, M.; Allen, T.; Levy, C.; White, D.E.; Mavroukakis, S.; Kreitman, R.J.; Rosenberg, S.A.; et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J. Immunol. 2007, 179, 4919–4928. [Google Scholar]
- Ashorn, P.; Moss, B.; Weinstein, J.N.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I.; Berger, E.A. Elimination of infectious human immunodeficiency virus from human T-cell cultures by synergistic action of CD4-Pseudomonas exotoxin and reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. USA 1990, 87, 8889–8893. [Google Scholar] [CrossRef]
- Bera, T.K.; Kennedy, P.E.; Berger, E.A.; Barbas, C.F., 3rd; Pastan, I. Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol. Med. 1998, 4, 384–391. [Google Scholar]
- Berger, E.A.; Pastan, I. Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog. 2010, 6, e1000803. [Google Scholar] [CrossRef]
- Li, H.; Gu, C.; Ren, Y.; Dai, Y.; Zhu, X.; Xu, J.; Li, Y.; Qiu, Z.; Zhu, J.; Zhu, Y.; et al. The efficacy of NP11-4-derived immunotoxin scFv-artesunate in reducing hepatic fibrosis induced by Schistosoma japonicum in mice. J. Biomed. Res. 2011, 25, 148–154. [Google Scholar] [CrossRef]
- Uckun, F.M.; Kersey, J.H.; Vallera, D.A.; Ledbetter, J.A.; Weisdorf, D.; Myers, D.E.; Haake, R.; Ramsay, N.K. Autologous bone marrow transplantation in high-risk remission T-lineage acute lymphoblastic leukemia using immunotoxins plus 4-hydroperoxycyclophosphamide for marrow purging. Blood 1990, 76, 1723–1733. [Google Scholar]
- Weisdorf, D.J.; Haake, R.; Miller, W.J.; McGlave, P.B.; LeBien, T.W.; Vallera, D.A.; Lasky, L.C.; Kim, T.H.; Peterson, B.A.; Ramsay, N.K.; et al. Autologous bone marrow transplantation for progressive non-Hodgkin’s lymphoma: Clinical impact of immunophenotype and in vitro purging. Bone Marrow Transplant. 1991, 8, 135–142. [Google Scholar]
- Kim, K.; Groman, N.B. Mode of inhibition of diphtheria toxin by ammonium chloride. J. Bacteriol. 1965, 90, 1557–1562. [Google Scholar]
- Draper, R.K.; Simon, M.I. The entry of diphtheria toxin into the mammalian cell cytoplasm: Evidence for lysosomal involvement. J. Cell Biol. 1980, 87, 849–854. [Google Scholar] [CrossRef]
- Sandvig, K.; Olsnes, S. Diphtheria toxin entry into cells is facilitated by low pH. J. Cell Biol. 1980, 87, 828–832. [Google Scholar] [CrossRef]
- Collier, R.J.; Gilliland, D.G.; Lory, S. Structure-activity relationships in diphtheria toxin and exotoxin A from Pseudomonas aeruginosa. Prog. Clin. Biol. Res. 1979, 31, 751–759. [Google Scholar]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar] [CrossRef]
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar] [CrossRef]
- Carreras-Sangra, N.; Alvarez-Garcia, E.; Herrero-Galan, E.; Tome, J.; Lacadena, J.; Alegre-Cebollada, J.; Onaderra, M.; Gavilanes, J.G.; Martinez-Del-Pozo, A. The therapeutic potential of fungal ribotoxins. Curr.Pharm.Biotechnol. 2008, 9, 153–160. [Google Scholar] [CrossRef]
- Zhan, H.; Choe, S.; Huynh, P.D.; Finkelstein, A.; Eisenberg, D.; Collier, R.J. Dynamic transitions of the transmembrane domain of diphtheria toxin: Disulfide trapping and fluorescence proximity studies. Biochemistry 1994, 33, 11254–11263. [Google Scholar] [CrossRef]
- Chaudhary, V.K.; Jinno, Y.; FitzGerald, D.; Pastan, I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc. Natl. Acad. Sci. USA 1990, 87, 308–312. [Google Scholar] [CrossRef]
- Simpson, J.C.; Dascher, C.; Roberts, L.M.; Lord, J.M.; Balch, W.E. Ricin cytotoxicity is sensitive to recycling between the endoplasmic reticulum and the Golgi complex. J. Biol. Chem. 1995, 270, 20078–20083. [Google Scholar]
- Wales, R.; Roberts, L.M.; Lord, J.M. Addition of an endoplasmic reticulum retrieval sequence to ricin A chain significantly increases its cytotoxicity to mammalian cells. J. Biol. Chem. 1993, 268, 23986–23990. [Google Scholar]
- Johnson, V.G.; Youle, R.J. A point mutation of proline 308 in diphtheria toxin B chain inhibits membrane translocation of toxin conjugates. J. Biol. Chem. 1989, 264, 17739–17744. [Google Scholar]
- Johnson, V.G.; Nicholls, P.J.; Habig, W.H.; Youle, R.J. The role of proline 345 in diphtheria toxin translocation. J. Biol. Chem. 1993, 268, 3514–3519. [Google Scholar]
- Ratts, R.; Zeng, H.; Berg, E.A.; Blue, C.; McComb, M.E.; Costello, C.E.; vanderSpek, J.C.; Murphy, J.R. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J. Cell Biol. 2003, 160, 1139–1150. [Google Scholar] [CrossRef]
- Antignani, A.; Youle, R.J. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Stone, M.; Amlot, P.; Fay, J.; May, R.; Till, M.; Newman, J.; Clark, P.; Collins, R.; Cunningham, D.; et al. Phase I immunotoxin trial in patients with B-cell lymphoma. Cancer Res. 1991, 51, 4052–4058. [Google Scholar]
- Lynch, T.J., Jr.; Lambert, J.M.; Coral, F.; Shefner, J.; Wen, P.; Blattler, W.A.; Collinson, A.R.; Ariniello, P.D.; Braman, G.; Cook, S.; et al. Immunotoxin therapy of small-cell lung cancer: A phase I study of N901-blocked ricin. J. Clin. Oncol. 1997, 15, 723–734. [Google Scholar]
- Ogata, M.; Chaudhary, V.K.; Pastan, I.; FitzGerald, D.J. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J. Biol. Chem. 1990, 265, 20678–20685. [Google Scholar]
- Gordon, V.M.; Klimpel, K.R.; Arora, N.; Henderson, M.A.; Leppla, S.H. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 1995, 63, 82–87. [Google Scholar]
- McKee, M.L.; FitzGerald, D.J. Reduction of furin-nicked Pseudomonas exotoxin A: An unfolding story. Biochemistry 1999, 38, 16507–16513. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Transport of protein toxins into cells: Pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002, 529, 49–53. [Google Scholar] [CrossRef]
- Sandvig, K.; Grimmer, S.; Iversen, T.G.; Rodal, K.; Torgersen, M.L.; Nicoziani, P.; van Deurs, B. Ricin transport into cells: Studies of endocytosis and intracellular transport. Int. J. Med. Microbiol. 2000, 290, 415–420. [Google Scholar] [CrossRef]
- Pawar, V.; De, A.; Briggs, L.; Omar, M.M.; Sweeney, S.T.; Lord, J.M.; Roberts, L.M.; Spooner, R.A.; Moffat, K.G. RNAi screening of Drosophila (Sophophora) melanogaster S2 cells for ricin sensitivity and resistance. J. Biomol. Screen. 2011, 16, 436–442. [Google Scholar] [CrossRef]
- Moreau, D.; Kumar, P.; Wang, S.C.; Chaumet, A.; Chew, S.Y.; Chevalley, H.; Bard, F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev. Cell 2011, 21, 231–244. [Google Scholar] [CrossRef]
- Wahome, P.G.; Bai, Y.; Neal, L.M.; Robertus, J.D.; Mantis, N.J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–323. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Derivation of specific antibody-producing tissue culture and tumor lines by cell fusion. Eur. J. Immunol. 1976, 6, 511–519. [Google Scholar] [CrossRef]
- Neville, D.M., Jr.; Srinivasachar, K.; Stone, R.; Scharff, J. Enhancement of immunotoxin efficacy by acid-cleavable cross-linking agents utilizing diphtheria toxin and toxin mutants. J. Biol. Chem. 1989, 264, 14653–14661. [Google Scholar]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.; Watson, G.J.; Knyba, R.E.; Wawrzynczak, E.J.; Blakey, D.C. New coupling agents for the synthesis of immunotoxins containing a hindered disulfide bond with improved stability in vivo. Cancer Res. 1987, 47, 5924–5931. [Google Scholar]
- Casellas, P.; Ravel, S.; Bourrie, B.J.; Derocq, J.M.; Jansen, F.K.; Laurent, G.; Gros, P. T-lymphocyte killing by T101-ricin A-chain immunotoxin: pH-dependent potentiation with lysosomotropic amines. Blood 1988, 72, 1197–1202. [Google Scholar]
- Hertler, A.A.; Frankel, A.E. Immunotoxins: A clinical review of their use in the treatment of malignancies. J. Clin. Oncol. 1989, 7, 1932–1942. [Google Scholar]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar]
- Chaudhary, V.K.; Queen, C.; Junghans, R.P.; Waldmann, T.A.; FitzGerald, D.J.; Pastan, I. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature 1989, 339, 394–397. [Google Scholar]
- Buchner, J.; Pastan, I.; Brinkmann, U. A method for increasing the yield of properly folded recombinant fusion proteins: Single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal. Biochem. 1992, 205, 263–270. [Google Scholar] [CrossRef]
- Brinkmann, U.; Buchner, J.; Pastan, I. Independent domain folding of Pseudomonas exotoxin and single-chain immunotoxins: Influence of interdomain connections. Proc. Natl. Acad. Sci. USA 1992, 89, 3075–3079. [Google Scholar] [CrossRef]
- Reiter, Y.; Brinkmann, U.; Kreitman, R.J.; Jung, S.H.; Lee, B.; Pastan, I. Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry 1994, 33, 5451–5459. [Google Scholar] [CrossRef]
- Brinkmann, U.; Reiter, Y.; Jung, S.H.; Lee, B.; Pastan, I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc. Natl. Acad. Sci. USA 1993, 90, 7538–7542. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Wayne, A.S.; Kreitman, R.J.; Pastan, I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer Res. 2011, 71, 6300–6309. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef]
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; FitzGerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800. [Google Scholar] [CrossRef]
- Onda, M.; Beers, R.; Xiang, L.; Lee, B.; Weldon, J.E.; Kreitman, R.J.; Pastan, I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc. Natl. Acad. Sci. USA 2011, 108, 5742–5747. [Google Scholar]
- Liu, W.; Onda, M.; Lee, B.; Kreitman, R.J.; Hassan, R.; Xiang, L.; Pastan, I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. USA 2012, 109, 11782–11787. [Google Scholar]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin-recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Pastan, I.; Chaudhary, V.; FitzGerald, D.J. Recombinant toxins as novel therapeutic agents. Annu. Rev. Biochem. 1992, 61, 331–354. [Google Scholar] [CrossRef]
- Laske, D.W.; Ilercil, O.; Akbasak, A.; Youle, R.J.; Oldfield, E.H. Efficacy of direct intratumoral therapy with targeted protein toxins for solid human gliomas in nude mice. J. Neurosurg. 1994, 80, 520–526. [Google Scholar] [CrossRef]
- Strom, T.B.; Anderson, P.L.; Rubin-Kelley, V.E.; Williams, D.P.; Kiyokawa, T.; Murphy, J.R. Immunotoxins and cytokine toxin fusion proteins. Semin. Immunol. 1990, 2, 467–479. [Google Scholar]
- Du, X.; Beers, R.; Fitzgerald, D.J.; Pastan, I. Differential cellular internalization of anti-CD19 and -CD22 immunotoxins results in different cytotoxic activity. Cancer Res. 2008, 68, 6300–6305. [Google Scholar] [CrossRef]
- Liu, X.F.; FitzGerald, D.J.; Pastan, I. The insulin receptor negatively regulates the action of Pseudomonas toxin-based immunotoxins and native Pseudomonas toxin. Cancer Res. 2013, 73, 2281–2288. [Google Scholar] [CrossRef]
- Wei, H.; Xiang, L.; Wayne, A.S.; Chertov, O.; FitzGerald, D.J.; Bera, T.K.; Pastan, I. Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. Proc. Natl. Acad. Sci. USA 2012, 109, 6898–6903. [Google Scholar]
- Mattoo, A.R.; FitzGerald, D.J. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int. J. Cancer 2013, 132, 978–987. [Google Scholar] [CrossRef]
- Zhang, Y.; Hansen, J.K.; Xiang, L.; Kawa, S.; Onda, M.; Ho, M.; Hassan, R.; Pastan, I. A flow cytometry method to quantitate internalized immunotoxins shows that taxol synergistically increases cellular immunotoxins uptake. Cancer Res. 2010, 70, 1082–1089. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, L.; Hassan, R.; Pastan, I. Immunotoxin and Taxol synergy results from a decrease in shed mesothelin levels in the extracellular space of tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 17099–17104. [Google Scholar] [CrossRef]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Cancer Res. 2002, 8, 3092–3099. [Google Scholar]
- Martin, P.J.; Hansen, J.A.; Vitetta, E.S. A ricin A chain-containing immunotoxin that kills human T lymphocytes in vitro. Blood 1985, 66, 908–912. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Blythman, H.E.; Casellas, P.; Frankel, A.E. An anti-CD5 immunotoxin for chronic lymphocytic leukemia: Enhancement of cytotoxicity with human serum albumin-monensin. Int. J. Cancer 1989, 43, 215–219. [Google Scholar] [CrossRef]
- Akiyama, S.; Seth, P.; Pirker, R.; FitzGerald, D.; Gottesman, M.M.; Pastan, I. Potentiation of cytotoxic activity of immunotoxins on cultured human cells. Cancer Res. 1985, 45, 1005–1007. [Google Scholar]
- FitzGerald, D.J.; Waldmann, T.A.; Willingham, M.C.; Pastan, I. Pseudomonas exotoxin-anti-TAC. Cell-specific immunotoxin active against cells expressing the human T cell growth factor receptor. J. Clin. Invest. 1984, 74, 966–971. [Google Scholar] [CrossRef]
- Wu, Y.N.; Gadina, M.; Tao Cheng, J.H.; Youle, R.J. Retinoic acid disrupts the Golgi apparatus and increases the cytosolic routing of specific protein toxins. J. Cell Biol. 1994, 125, 743–753. [Google Scholar] [CrossRef]
- Fitzgerald, D.J.; Moskatel, E.; Ben-Josef, G.; Traini, R.; Tendler, T.; Sharma, A.; Antignani, A.; Mussai, F.; Wayne, A.; Kreitman, R.J.; et al. Enhancing immunotoxin cell-killing activity via combination therapy with ABT-737. Leuk. Lymphoma. 2011, 52, 79–81. [Google Scholar]
- Traini, R.; Ben-Josef, G.; Pastrana, D.V.; Moskatel, E.; Sharma, A.K.; Antignani, A.; Fitzgerald, D.J. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of Pseudomonas exotoxin-based proteins to the cell cytosol. Mol. Cancer Ther. 2010, 9, 2007–2015. [Google Scholar] [CrossRef]
- Risberg, K.; Fodstad, O.; Andersson, Y. Synergistic anticancer effects of the 9.2.27PE immunotoxin and ABT-737 in melanoma. PLoS One 2011, 6, 24012. [Google Scholar]
- Du, X.; Xiang, L.; Mackall, C.; Pastan, I. Killing of resistant cancer cells with low Bak by a combination of an antimesothelin immunotoxin and a TRAIL Receptor 2 agonist antibody. Clin. Cancer Res. 2011, 17, 5926–5934. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, L.; Hassan, R.; Paik, C.H.; Carrasquillo, J.A.; Jang, B.S.; Le, N.; Ho, M.; Pastan, I. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin. Cancer Res. 2006, 12, 4695–4701. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antignani, A.; FitzGerald, D. Immunotoxins: The Role of the Toxin. Toxins 2013, 5, 1486-1502. https://doi.org/10.3390/toxins5081486
Antignani A, FitzGerald D. Immunotoxins: The Role of the Toxin. Toxins. 2013; 5(8):1486-1502. https://doi.org/10.3390/toxins5081486
Chicago/Turabian StyleAntignani, Antonella, and David FitzGerald. 2013. "Immunotoxins: The Role of the Toxin" Toxins 5, no. 8: 1486-1502. https://doi.org/10.3390/toxins5081486
APA StyleAntignani, A., & FitzGerald, D. (2013). Immunotoxins: The Role of the Toxin. Toxins, 5(8), 1486-1502. https://doi.org/10.3390/toxins5081486