Atractaspis aterrima Toxins: The First Insight into the Molecular Evolution of Venom in Side-Stabbers


Abstract
:1. Introduction
2. Results
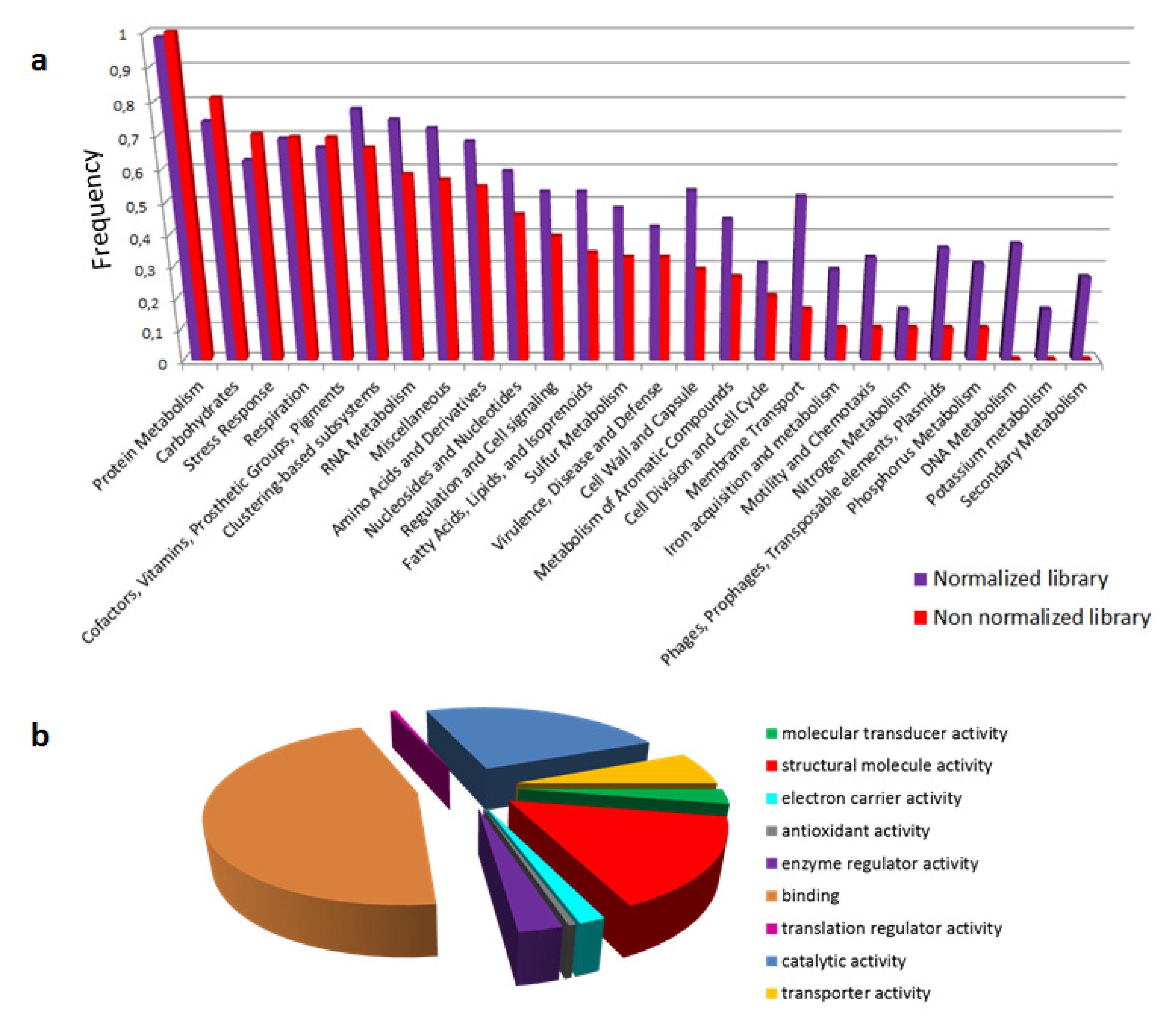
2.1. Sequencing and Assembly Statistics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normalized | Non-normalized | |
|---|---|---|
| Sequencing | ||
| Total Number of Reads | 724,119 | 581,370 |
| Total Number of Bases | 249,123,133 | 183,358,305 |
| Average Read Length | 344 | 315 |
| Assembly Results | ||
| Number Assembled | 427,470 | 266,199 |
| Number tooshort | 27,744 | 0 |
| Sum of Large Contigs (>1 Kb) | ||
| Total number of reads | 86,119 | 35,356 |
| Number of Large Contigs | 2197 | 265 |
| Total number of bases | 2,914,941 | 344,247 |
| Sum of All Contigs | ||
| Total number of reads | 427,470 | 266,199 |
| Number of All Contigs | 69,975 | 57,962 |
| Total number of bases | 35,504,970 | 22,851,131 |
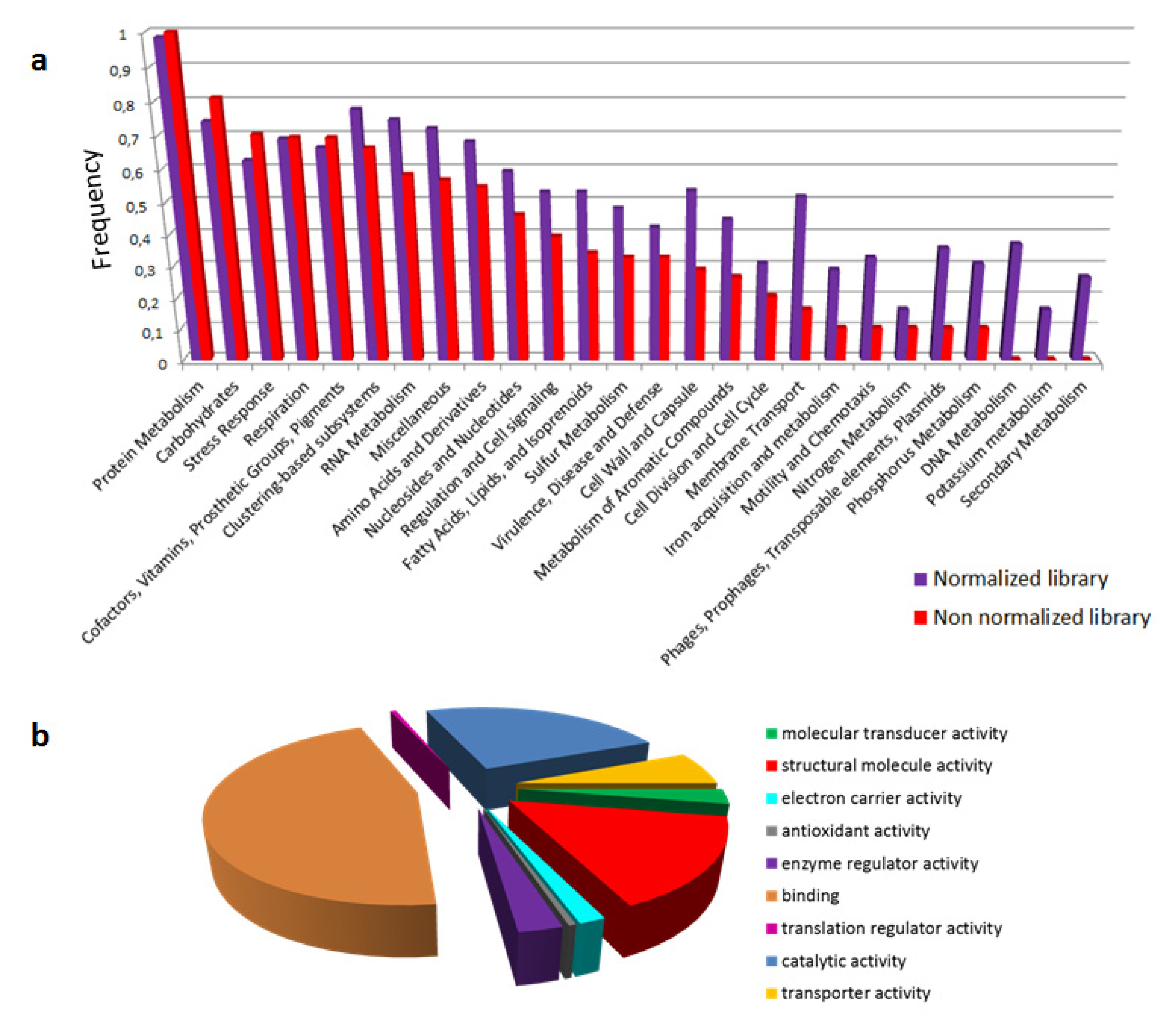
2.2. Functional Annotation of Atractaspis aterrima Venom Gland Transcriptome
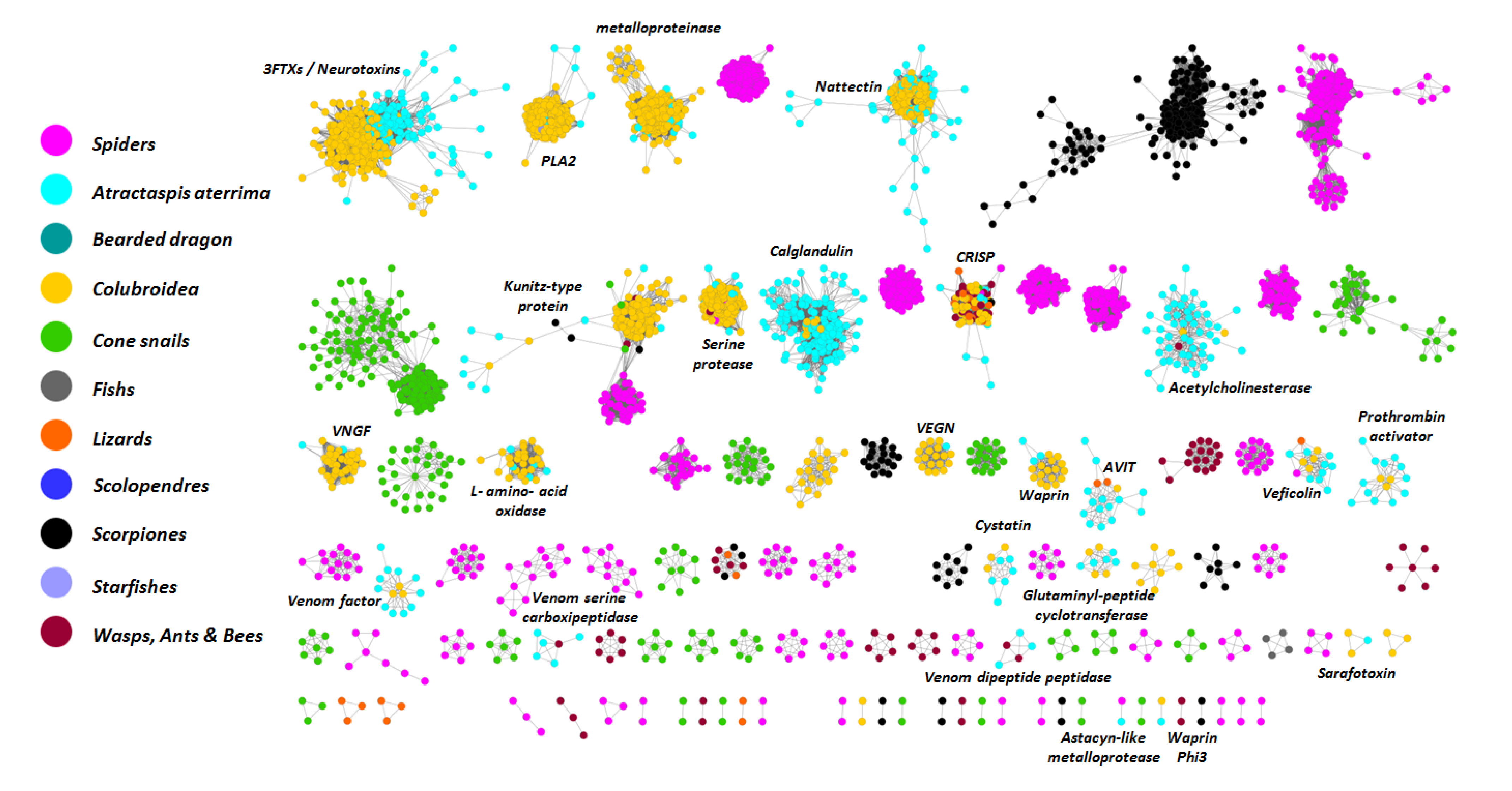
2.3. Analysis of Toxin Transcripts
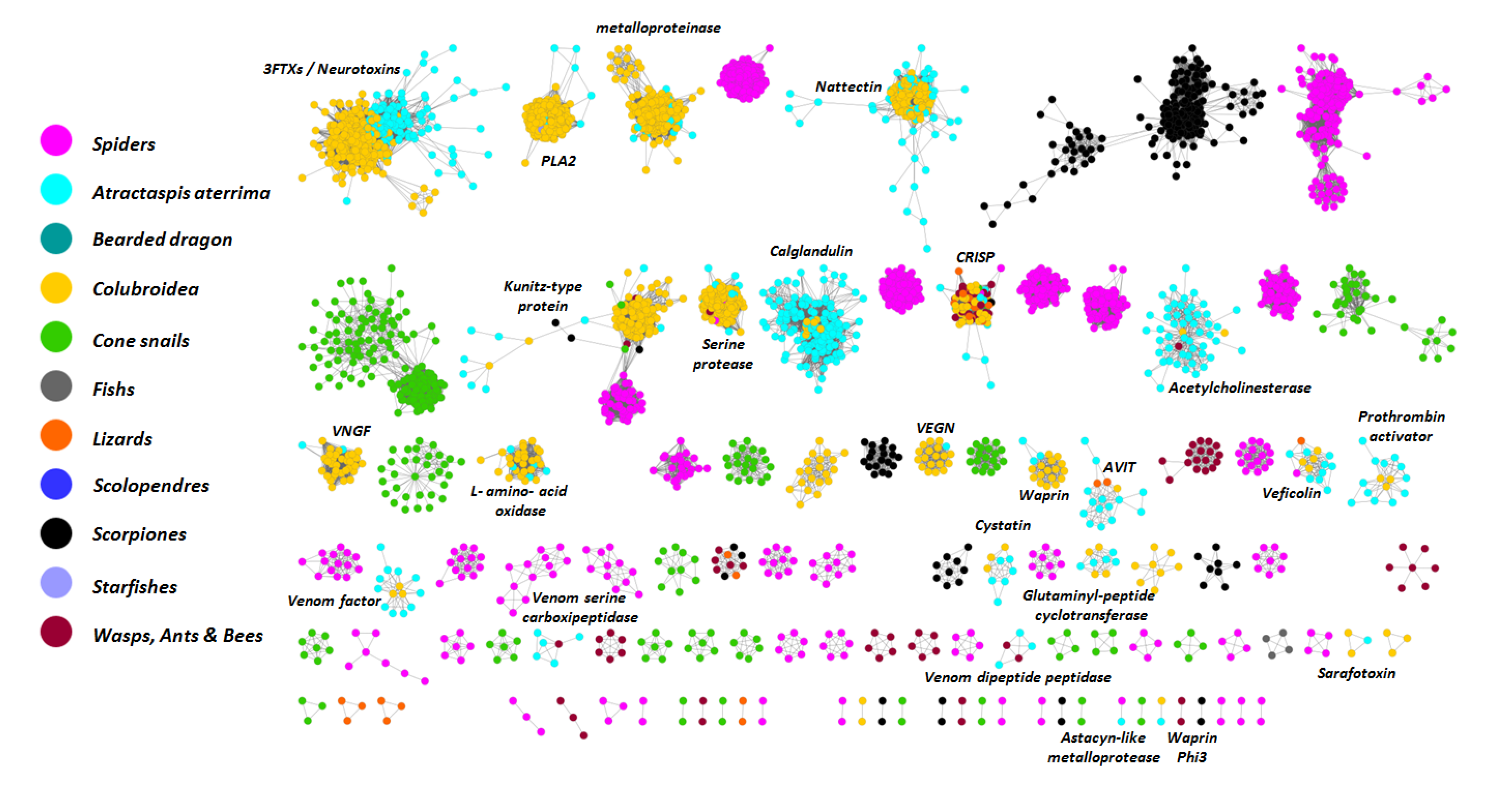
| Toxin family | Isoform(s) | Remarkable feature |
|---|---|---|
| Three finger toxin | 13 | Large diversity |
| Kunitz type/TFPI | 3 | Original signal peptide/one Kunitz type domain |
| AVIT | 3 | Distantly related to dendroapsis and varanus AVITs |
| Choline esterase | 6 | Original signal peptide |
| Crotamine | 1 | Conserved signal peptide (Crotalus genus), conserved cysteine pattern but low similarity through mature peptide |
| Metaloproteinase disintegrin (ADAM) | 1 | Truncated sequence |
| C-type lectin | 4 | Two distinct groups |
| Waprin | 6 | None |
| Kallikrein / Serine protease | 2 | None |
| CRISP | 1 | Highly similar to Latisemin toxin from Laticauda semifasciata |
| Venom nerve Growth Factor (VNGF) | 1 | Partial sequence nearly identical to viperidae’s VNGFs |
| Lipocalin | 3 | Two different groups. Major compound of the venom gland’s transcriptome |
| PLA2 | 1 | Partial sequence. Highly similar to phospholipase A2 type II from Leioheterodon madagascariensis |
| Cystacin | 4 | Partial sequences highly similar to Crotalus adamanteus toxins but lack signal peptide |
| Sarafotoxin | 2 | Partial sequence. Matching only two reads from the normalized library |
| Calglandulines | 1 | 95% identical to the Elipadae Austrelaps superbus caglandulin sequence |
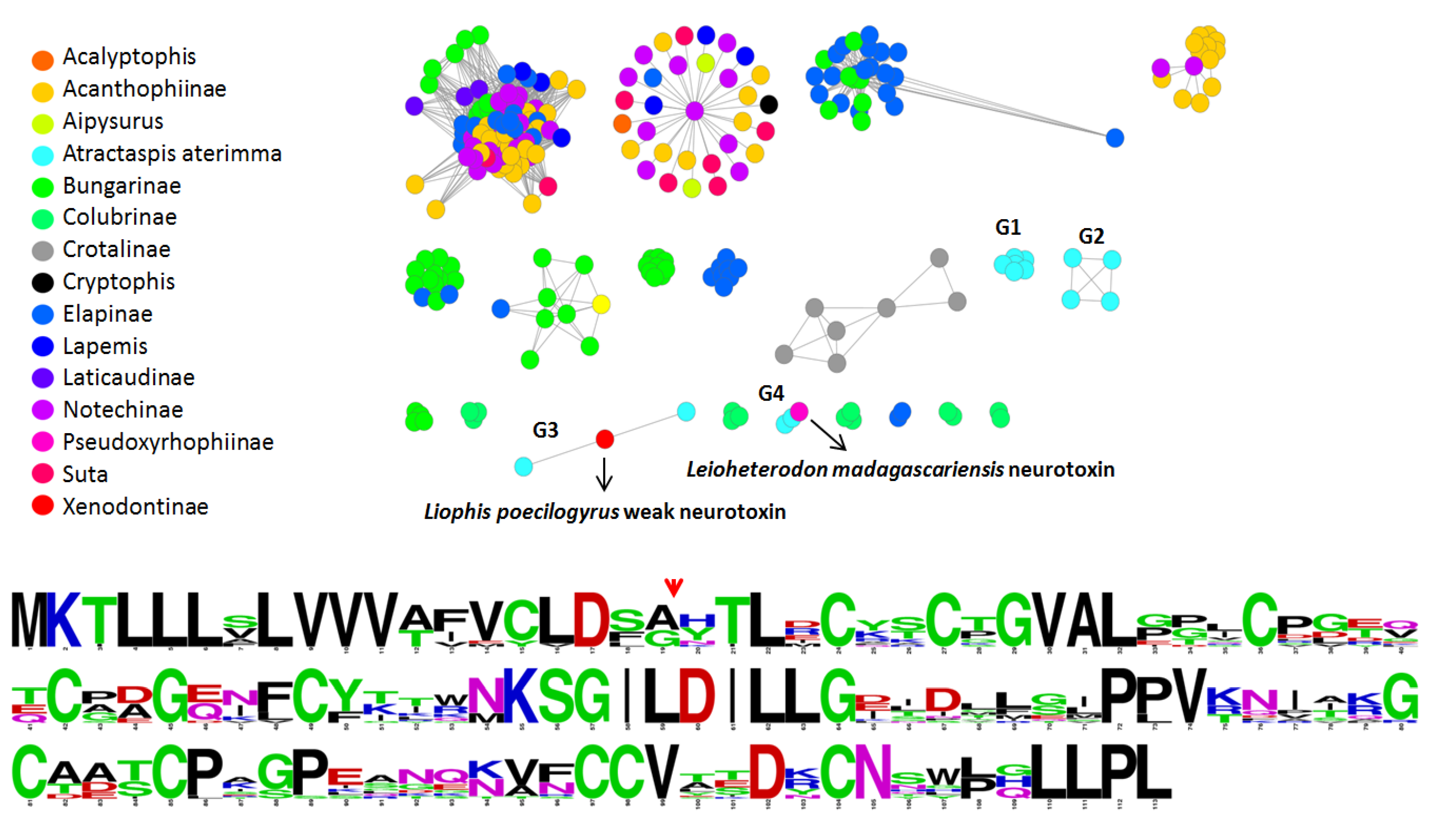
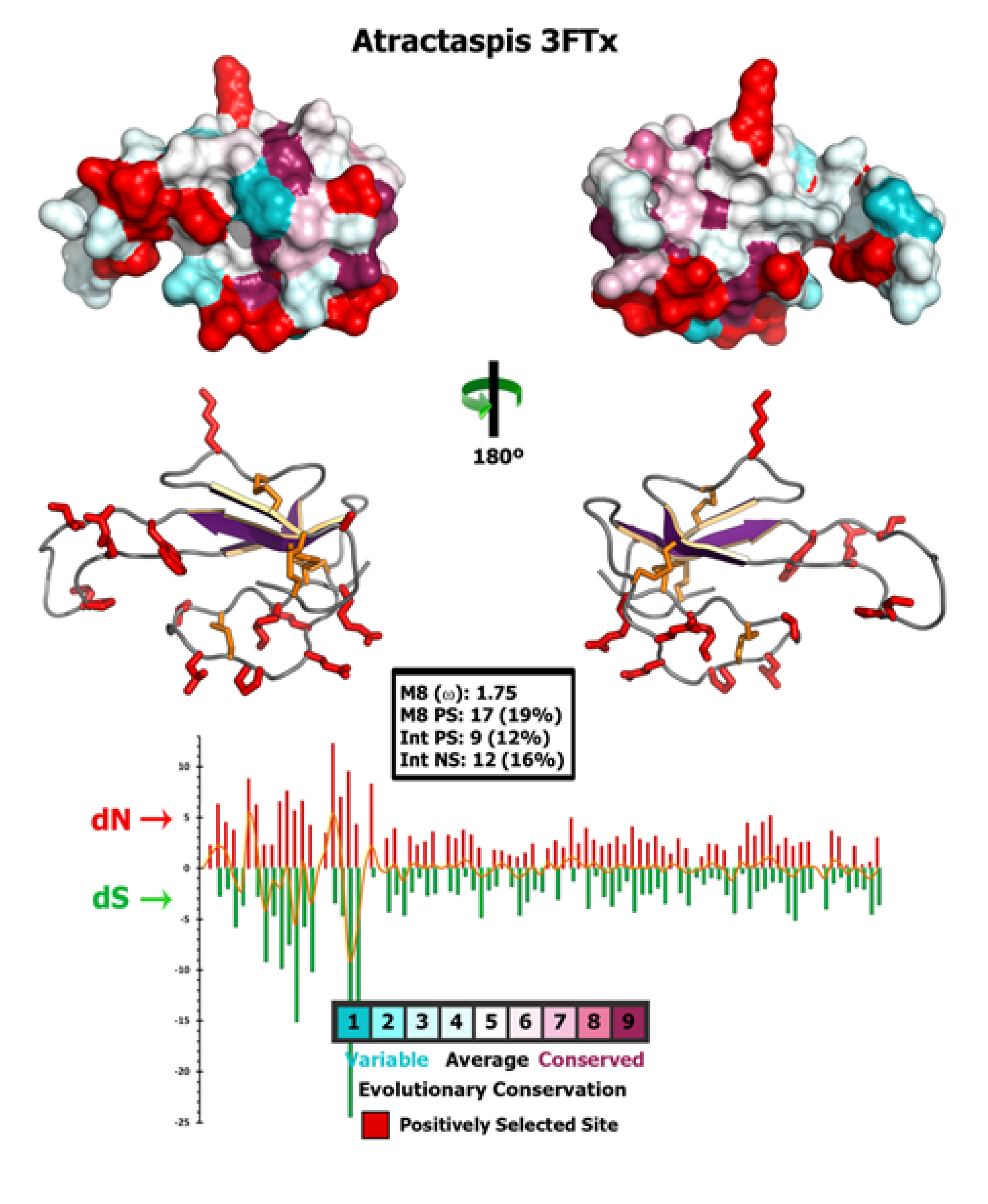
2.4. Molecular Evolution of Atractaspis aterrima Three-Finger Toxins


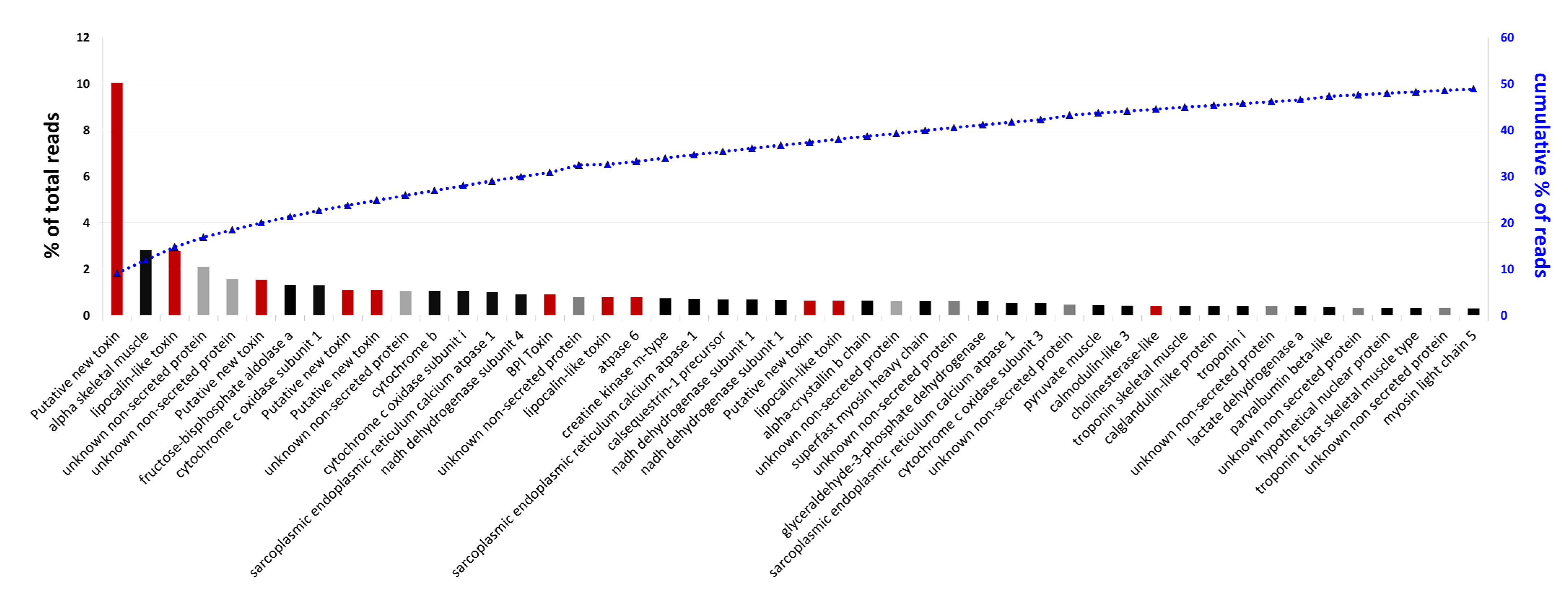
2.5. Analysis of Highly Expressed Transcripts and Detection of Unknown Proteins
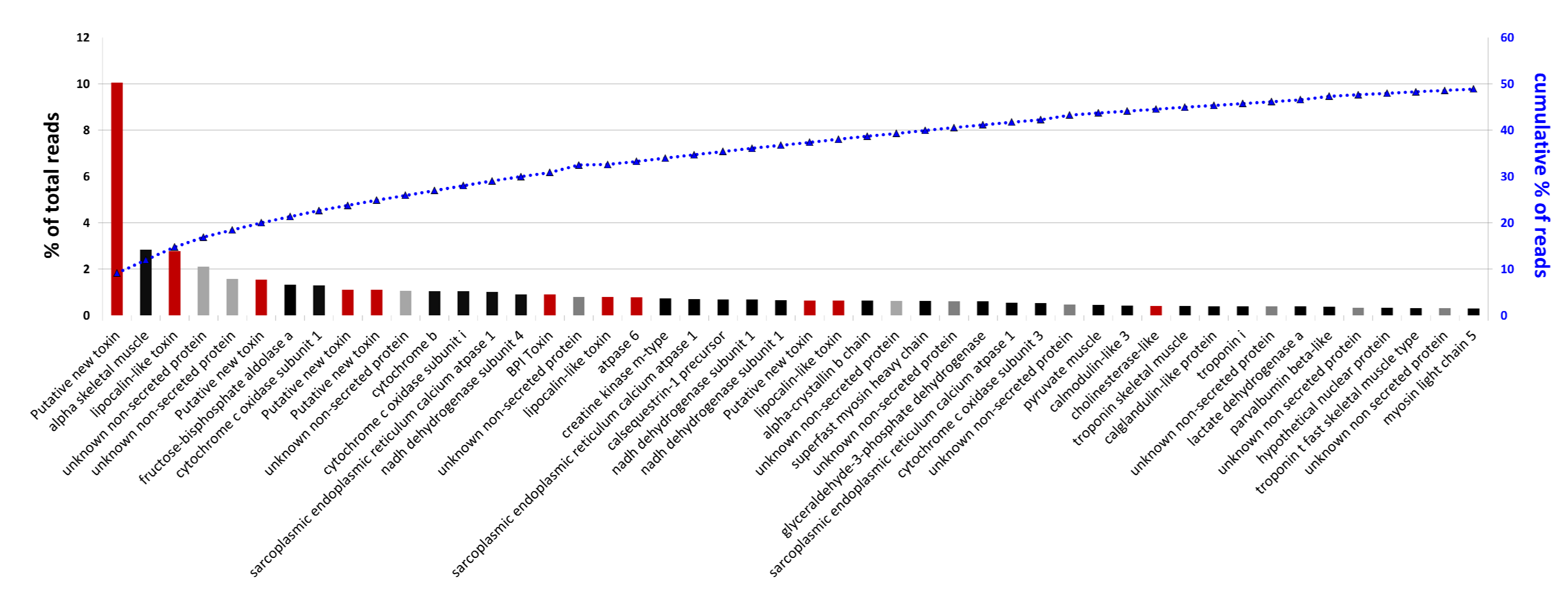
3. Experimental Section
3.1. Snake Venom Gland cDNA Synthesis and Sequencing
3.2. Bioinformatic Processing of the 454 Reads and Annotation of the Dataset
3.3. Functional Annotation of Contigs
3.4. Prediction of Non-Matching Sequences
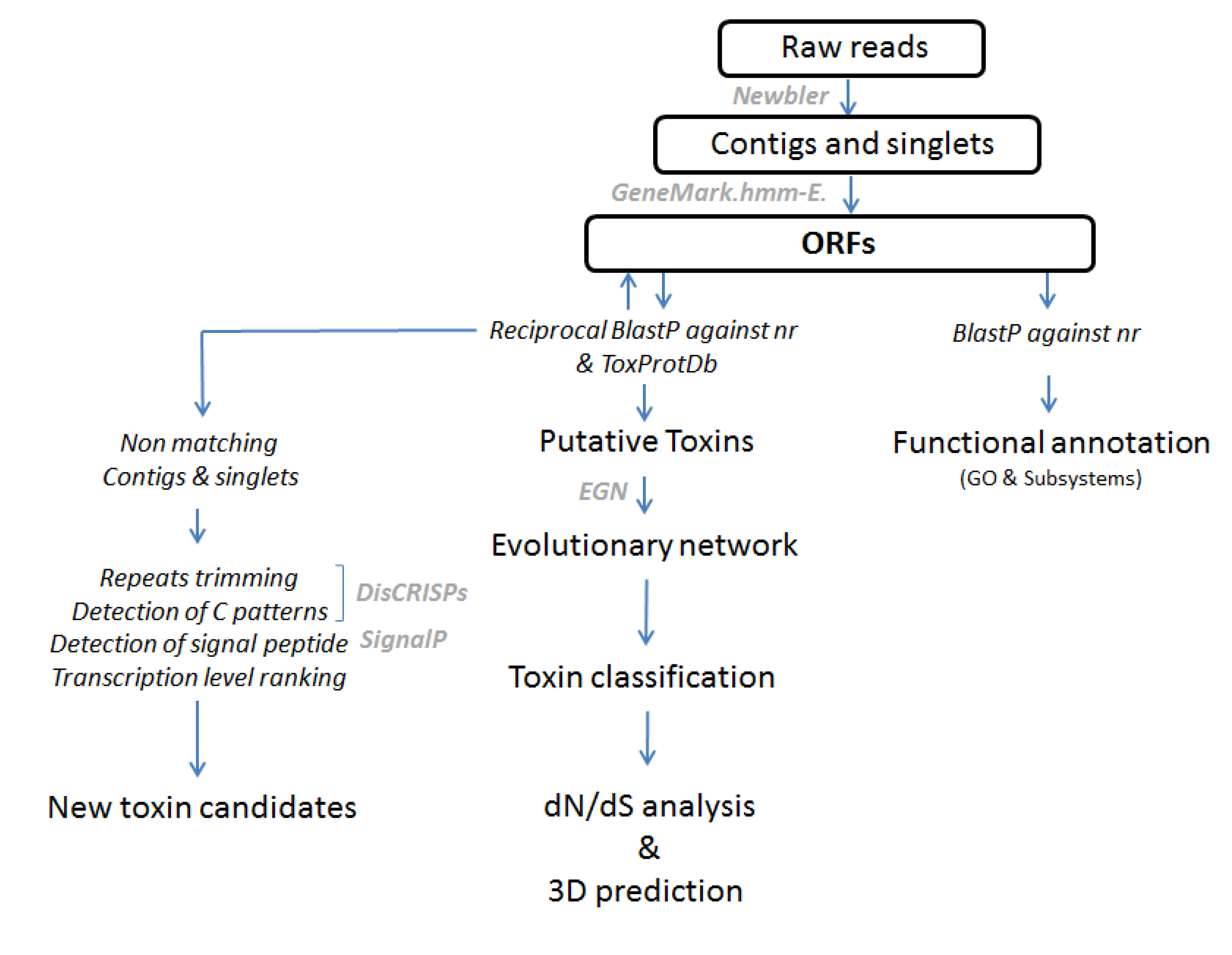
3.5. Diversity Analysis Using Evolutionary Networks
3.6. Selection Analyses
3.7. Structural Analyses
4. Conclusion
Acknowledgments
Conflicts of interest
References
- Fry, B.G.; Casewell, N.R.; Wüster, W.; Vidal, N.; Young, B.; Jackson, T.N. The structural and functional diversification of the Toxicofera reptile venom system. Toxicon 2012, 60, 434–448. [Google Scholar] [CrossRef]
- Ducancel, F. Endothelin-like peptides. Cell. Mol. Life Sci. 2005, 62, 2828–2839. [Google Scholar] [CrossRef]
- Kochva, E. Atractaspis (serpentes, Atractaspididae) the burrowing asp. A mulstidisciplinary mini review. Bull. Nat. Hist. Mus. Zool. 2002, 68, 91–99. [Google Scholar]
- Golani, I.; Kochva, E. Biting behaviour of Atractaspis. Copeia 1988, 792–797. [Google Scholar] [CrossRef]
- Weiser, E.; Wollberg, Z.; Kochva, E.; Lee, S.Y. Cardiotoxic effects of the venom of the burrowing asp, Atractaspis engaddensis (Atractaspididae, Ophidia). Toxicon 1984, 22, 767–774. [Google Scholar] [CrossRef]
- Kurnik, D.; Haviv, Y.; Kochva, E. A snake bite by the Burrowing Asp, Atractaspis engaddensis. Toxicon 1999, 37, 223–227. [Google Scholar] [CrossRef]
- Kloog, Y.; Ambar, I.; Sokolovsky, M.; Kochva, E.; Wollberg, Z.; Bdolah, A. Sarafotoxin, a novel vasoconstrictor peptide: Phosphoinositide hydrolysis in rat heart and brain. Science 1988, 242, 268–270. [Google Scholar]
- Ducancel, F.; Matre, V.; Dupont, C.; Lajeunesse, E.; Wollberg, Z.; Bdolah, A.; Kochva, E.; Boulain, J.C.; Ménez, A. Cloning and sequence analysis of cDNAs encoding precursors of sarafotoxins. Evidence for an unusual “rosary-type” organization. J. Biol. Chem. 1993, 268, 3052–3055. [Google Scholar]
- sHayashi, M.A.F.; Ligny-Lemaire, C.; Wollberg, Z.; Wery, M.; Galat, A.; Ogawa, T.; Muller, B.H.; Lamthanh, H.; Doljansky, Y.; Bdolah, A.; et al. Long-sarafotoxins: Characterization of a new family of endothelin-like peptides. Peptides 2004, 25, 1243–1251. [Google Scholar] [CrossRef]
- Quinton, L.; Le Caer, J.-P.; Phan, G.; Ligny-Lemaire, C.; Bourdais-Jomaron, J.; Ducancel, F.; Chamot-Rooke, J. Characterization of toxins within crude venoms by combined use of Fourier transform mass spectrometry and cloning. Anal. Chem. 2005, 77, 6630–6639. [Google Scholar] [CrossRef]
- Kochva, E.; Bdolah, A.; Wollberg, Z. Sarafotoxins and endothelins: Evolution, structure and function. Toxicon 1993, 31, 541–568. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.A.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef]
- Fry, B.G. From genome to evenome”enMolecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 2005, 15, 403–420. [Google Scholar] [CrossRef]
- Fry, B.G.; Undheim, E.A.B.; Ali, S.A.; Jackson, T.N.W.; Debono, J.; Scheib, H.; Ruder, T.; Morgenstern, D.; Cadwallader, L.; Whitehead, D.; et al. Squeezers and leaf-cutters: Differential diversification and degeneration of the venom system in toxicoferan reptiles. Mol. Cell. Proteomics 2013, 12, 1881–1899. [Google Scholar] [CrossRef]
- Mourier, G.; Hajj, M.; Cordier, F.; Zorba, A.; Gao, X.; Coskun, T.; Herbet, A.; Marcon, E.; Beau, F.; Delepierre, M.; et al. Pharmacological and structural characterization of long-sarafotoxins, a new family of endothelin-like peptides: Role of the C-terminus extension. Biochimie 2012, 94, 461–470. [Google Scholar] [CrossRef]
- Fry, B.G.; Scheib, H.; van der Weerd, L.; Young, B.; McNaughtan, J.; Ramjan, S.F.R.; Vidal, N.; Poelmann, R.E.; Norman, J.A. Evolution of an arsenal: Structural and functional diversification of the venom system in the advanced snakes (Caenophidia). Mol. Cell. Proteomics MCP 2008, 7, 215–246. [Google Scholar]
- Fry, B.G.; Scheib, H.; de L M Junqueira de Azevedo, I.; Silva, D.A.; Casewell, N.R. Novel transcripts in the maxillary venom glands of advanced snakes. Toxicon 2012, 59, 696–708. [Google Scholar] [CrossRef]
- Derrien, T.; Guigi, R.; Johnson, R. The long non-coding RNAs: A new (P)layer in the ydark matterh. Front. Genet. 2012, 1. [Google Scholar] [CrossRef]
- Terrat, Y.; Biass, D.; Dutertre, S.; Favreau, P.; Remm, M.; Stöcklin, R.; Piquemal, D.; Ducancel, F. High-resolution picture of a venom gland transcriptome: Case study with the marine snail Conus consors. Toxicon 2012, 59, 34–46. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell. Proteomics 2013, 12, 312–329. [Google Scholar] [CrossRef]
- Becker, A.; Dowdle, E.B.; Hechler, U.; Kauser, K.; Donner, P.; Schleuning, W.D. Bibrotoxin, a novel member of the endothelin/sarafotoxin peptide family, from the venom of the burrowing asp Atractaspis bibroni. FEBS Lett. 1993, 315, 100–103. [Google Scholar] [CrossRef]
- Fry, B.G.; Wüster, W.; Kini, R.M.; Brusic, V.; Khan, A.; Venkataraman, D.; Rooney, A.P. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 2003, 57, 110–129. [Google Scholar] [CrossRef]
- Fry, B.G.; Lumsden, N.G.; Wüster, W.; Wickramaratna, J.C.; Hodgson, W.C.; Kini, R.M. Isolation of a neurotoxin (alpha-colubritoxin) from a nonvenomous colubrid: Evidence for early origin of venom in snakes. J. Mol. Evol. 2003, 57, 446–452. [Google Scholar] [CrossRef]
- Borodovsky, M.; Lomsadze, A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinformatics 2011. [Google Scholar] [CrossRef]
- Durban, J.; Juárez, P.; Angulo, Y.; Lomonte, B.; Flores-Diaz, M.; Alape-Giraz, A.; Sasa, M.; Sanz, L.; Gutiérrez, J.M.; Dopazo, J.; et al. Profiling the venom gland transcriptomes of Costa Rican snakes by 454 pyrosequencing. BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Halary, S.; Leigh, J.W.; Cheaib, B.; Lopez, P.; Bapteste, E. Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. USA 2010, 107, 127–132. [Google Scholar]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar]
- Pond, S.L.K.; Scheffler, K.; Gravenor, M.B.; Poon, A.F.Y.; Frost, S.D.W. Evolutionary fingerprinting of genes. Mol. Biol. Evol. 2010, 27, 520–536. [Google Scholar]
- Kosakovsky Pond, S.L.; Murrell, B.; Fourment, M.; Frost, S.D.W.; Delport, W.; Scheffler, K. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 2011, 28, 3033–3043. [Google Scholar]
- Kelley, L.A.; Sternberg, M.J.E. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef]
- DeLano, WL. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Armon, A.; Graur, D.; Ben-Tal, N. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001, 307, 447–463. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
- Trevisan-Silva, D.; Gremski, L.H.; Chaim, O.M.; da Silveira, R.B.; Meissner, G.O.; Mangili, O.C.; Barbaro, K.C.; Gremski, W.; Veiga, S.S.; Senff-Ribeiro, A. Astacin-like metalloproteases are a gene family of toxins present in the venom of different species of the brown spider (genus Loxosceles). Biochimie 2010, 92, 21–32. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Yasumasu, S.; Shimizu, A.; Hiroi, J.; Yoshizaki, N.; Nagata, K.; Tanokura, M.; Iuchi, I. Purification and gene cloning of Fundulus heteroclitus hatching enzyme. A hatching enzyme system composed of high choriolytic enzyme and low choriolytic enzyme is conserved between two different teleosts, Fundulus heteroclitus and medaka Oryzias latipes. FEBS J. 2005, 272, 4315–4326. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Terrat, Y.; Sunagar, K.; Fry, B.G.; Jackson, T.N.W.; Scheib, H.; Fourmy, R.; Verdenaud, M.; Blanchet, G.; Antunes, A.; Ducancel, F. Atractaspis aterrima Toxins: The First Insight into the Molecular Evolution of Venom in Side-Stabbers. Toxins 2013, 5, 1948-1964. https://doi.org/10.3390/toxins5111948
Terrat Y, Sunagar K, Fry BG, Jackson TNW, Scheib H, Fourmy R, Verdenaud M, Blanchet G, Antunes A, Ducancel F. Atractaspis aterrima Toxins: The First Insight into the Molecular Evolution of Venom in Side-Stabbers. Toxins. 2013; 5(11):1948-1964. https://doi.org/10.3390/toxins5111948
Chicago/Turabian StyleTerrat, Yves, Kartik Sunagar, Bryan G. Fry, Timothy N. W. Jackson, Holger Scheib, Rudy Fourmy, Marion Verdenaud, Guillaume Blanchet, Agostinho Antunes, and Frederic Ducancel. 2013. "Atractaspis aterrima Toxins: The First Insight into the Molecular Evolution of Venom in Side-Stabbers" Toxins 5, no. 11: 1948-1964. https://doi.org/10.3390/toxins5111948
APA StyleTerrat, Y., Sunagar, K., Fry, B. G., Jackson, T. N. W., Scheib, H., Fourmy, R., Verdenaud, M., Blanchet, G., Antunes, A., & Ducancel, F. (2013). Atractaspis aterrima Toxins: The First Insight into the Molecular Evolution of Venom in Side-Stabbers. Toxins, 5(11), 1948-1964. https://doi.org/10.3390/toxins5111948