1H NMR Spectroscopy-Based Metabolomic Assessment of Uremic Toxicity, with Toxicological Outcomes, in Male Rats Following an Acute, Mid-Life Insult from Ochratoxin A

Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiment
2.2. Histopathology
2.3. Urinalysis
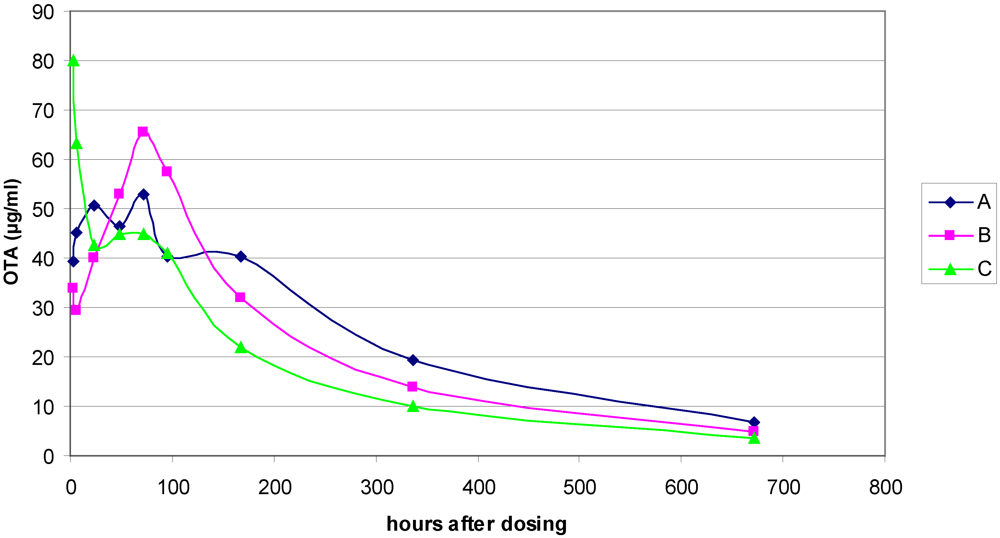
2.4. Analysis of Ochratoxin A in Rat Plasma
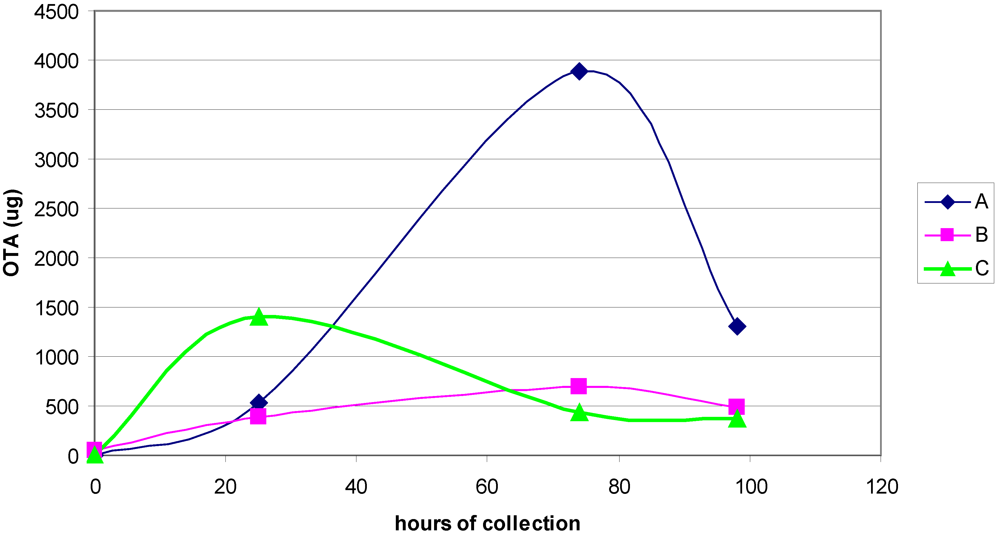
2.5. Measurement of Ochratoxin A in Rat Urine
2.6. Measurement of Ochratoxin Alpha in Rat Urine
2.7. 1H NMR Spectroscopic Analysis of Whole Rat Urine
2.8. Multivariate Statistical Analysis of 1H NMR Spectral Data
3. Results
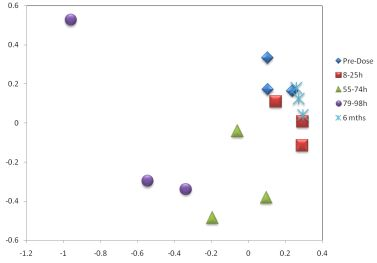
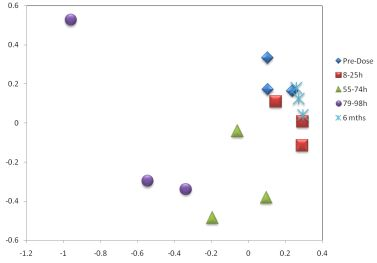
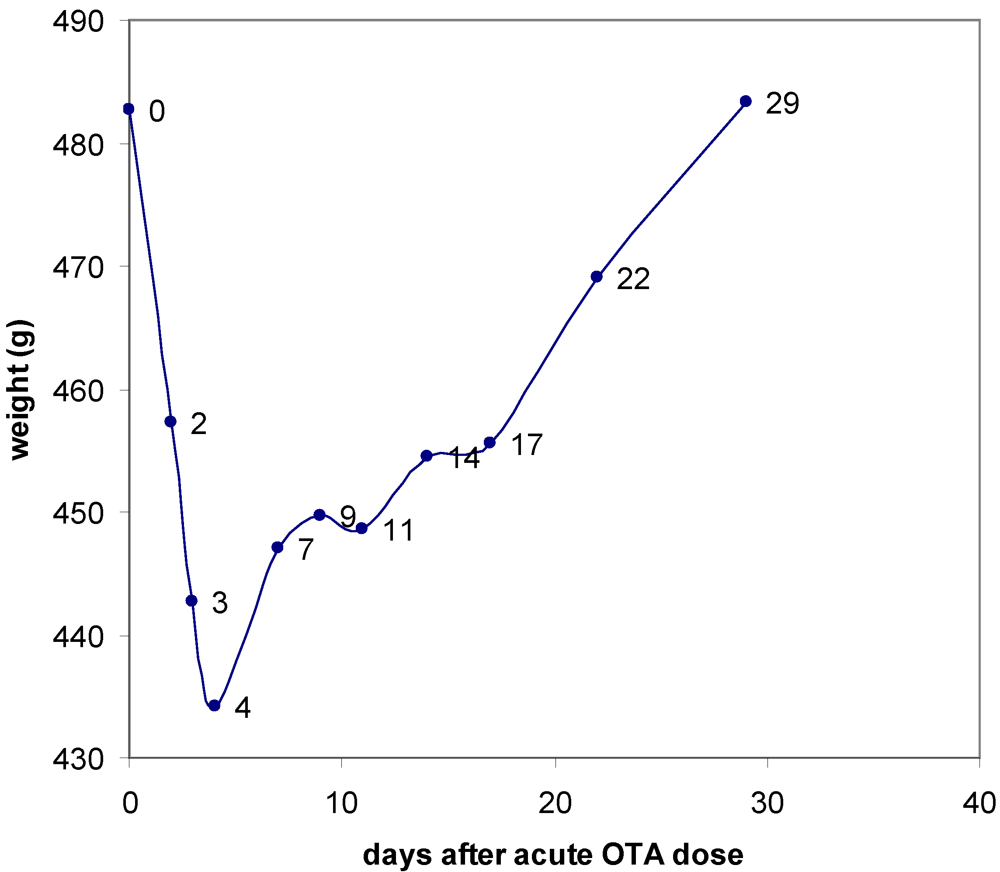
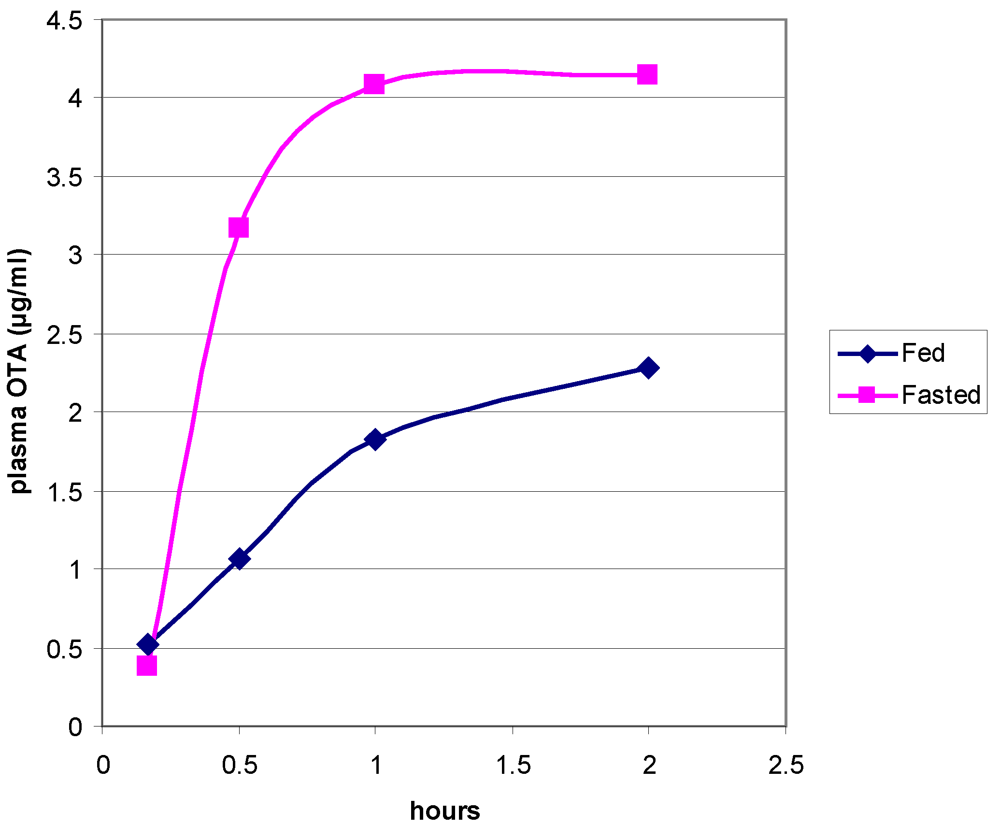
3.1. The Acute Toxicity Phase
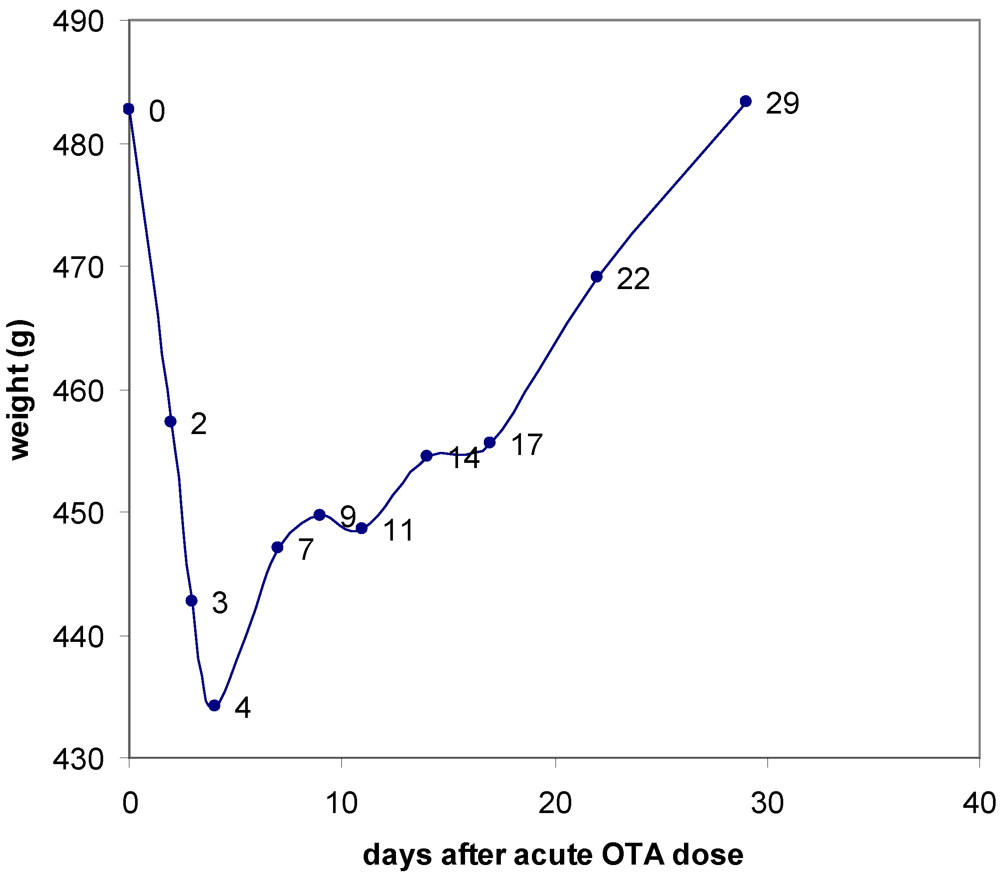
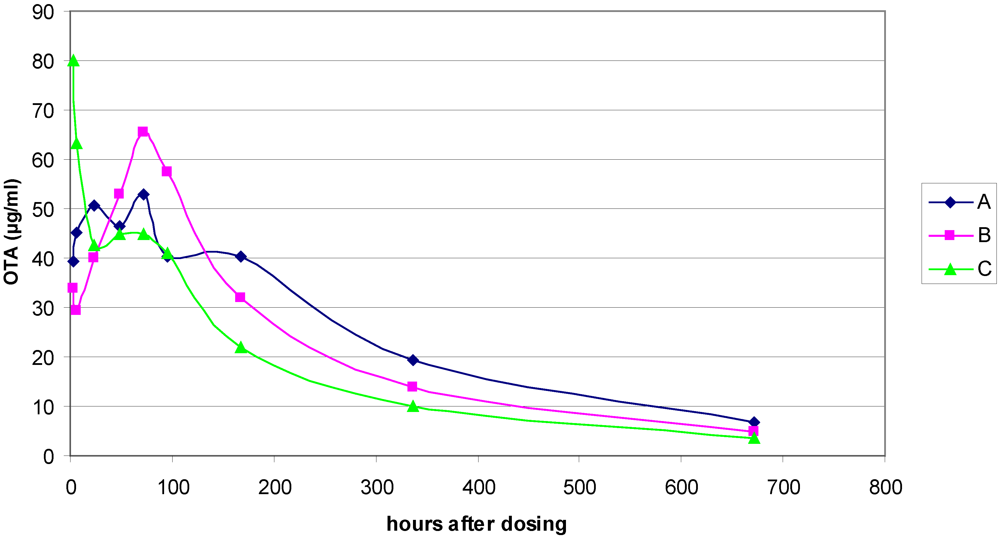

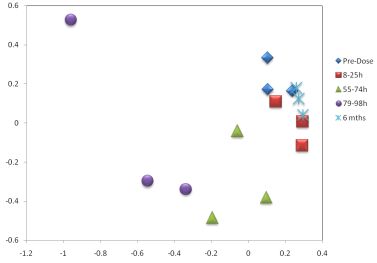{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Volumes | Creatinine | Calcium | Urate | Protein | Na | K | PO4 | Urea | |
|---|---|---|---|---|---|---|---|---|---|
| Pre-OTA | 7.5 (2.60) | 10.63 (2.65) | 0.31 (0.02) | 0.12 (0.04) | 0.40 (0.16) | 7.54 (3.18) | 19.45 (4.62) | 2.28 (0.77) | 87.39 (6.79) |
| 8–25 h | 7.17 (1.04) | 9.18 (0.98) | 0.33 (0.09) | 0.2 (0.05) | 0.39 (0.16) | 12.32 (4.77) | 12.83 (2.03) | 3.55 (1.40) | 77.27 (0.67) |
| 55–74 h | 15.8 (10.37) | 6.50 (3.94) | 0.18 (0.11) | 0.2 (0.15) | 0.42 (0.12) | 11.67 (2.24) | 12.66 (0.82) | 5.56 (0.68) | 60.14 (2.02) |
| 79–88 h | 30.7 (2.87) | 2.29 (0.13) | 0.13 (0.09) | 0.1 (0.02) | 0.63 (0.07) | 7.03 (1.79) | 18.76 (2.05) | 4.41 (1.02) | 58.00 (1.82) |
| 6 months | 7.97 (1.56) | 10.55 (1.96) | 0.27 (0.11) | 0.17 (0.02) | 0.37 (0.23) | 3.39 (1.20) | 14.93 (1.46) | 2.64 (0.40) | 61.44 (1.64) |
| 12 months | 14.5 | 4.66 | 1.28 | 0.34 | 3.04 | 10.33 | 22.27 | 6.19 | 52.56 |

3.2. Survival and Necropsy
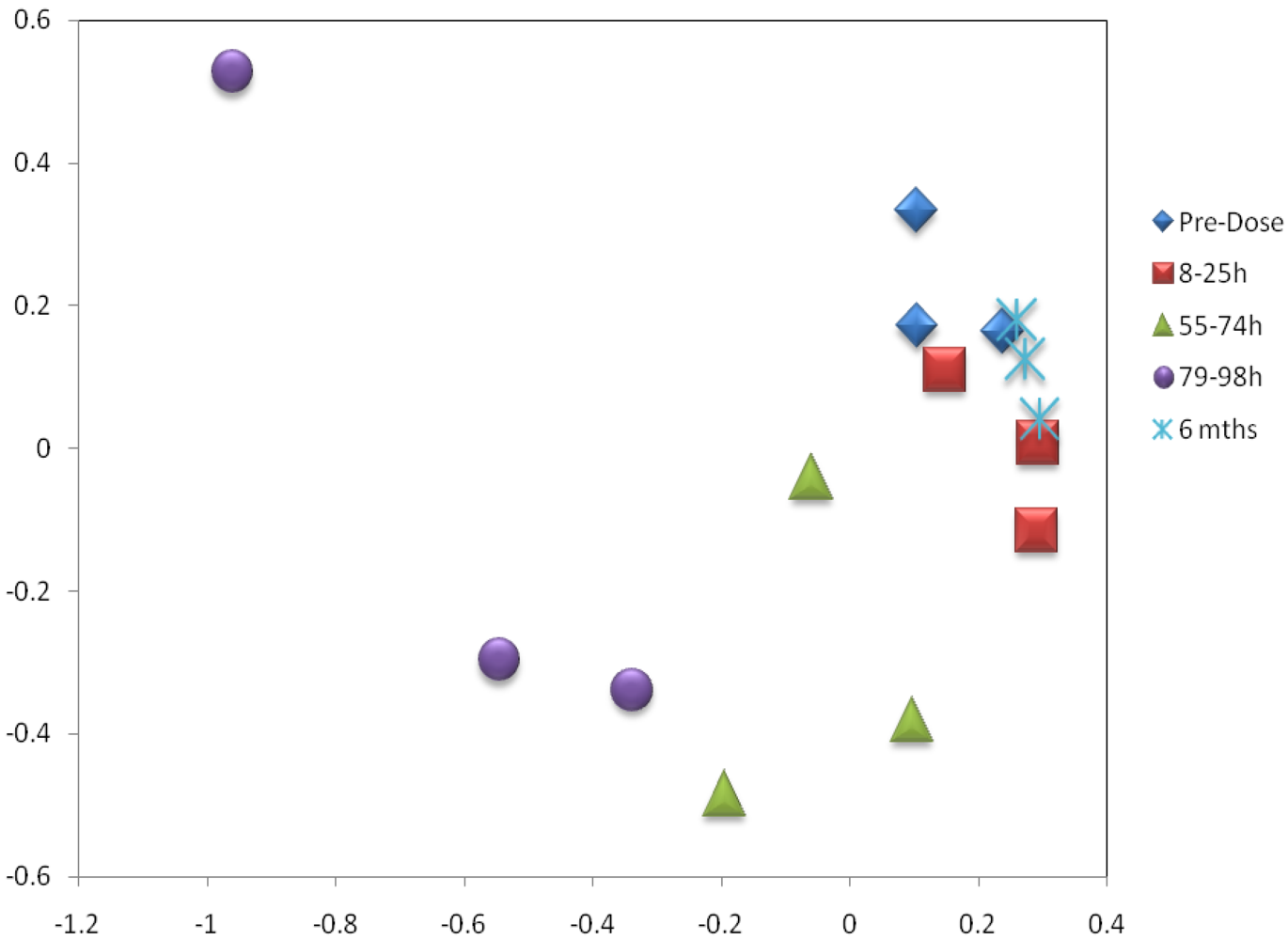
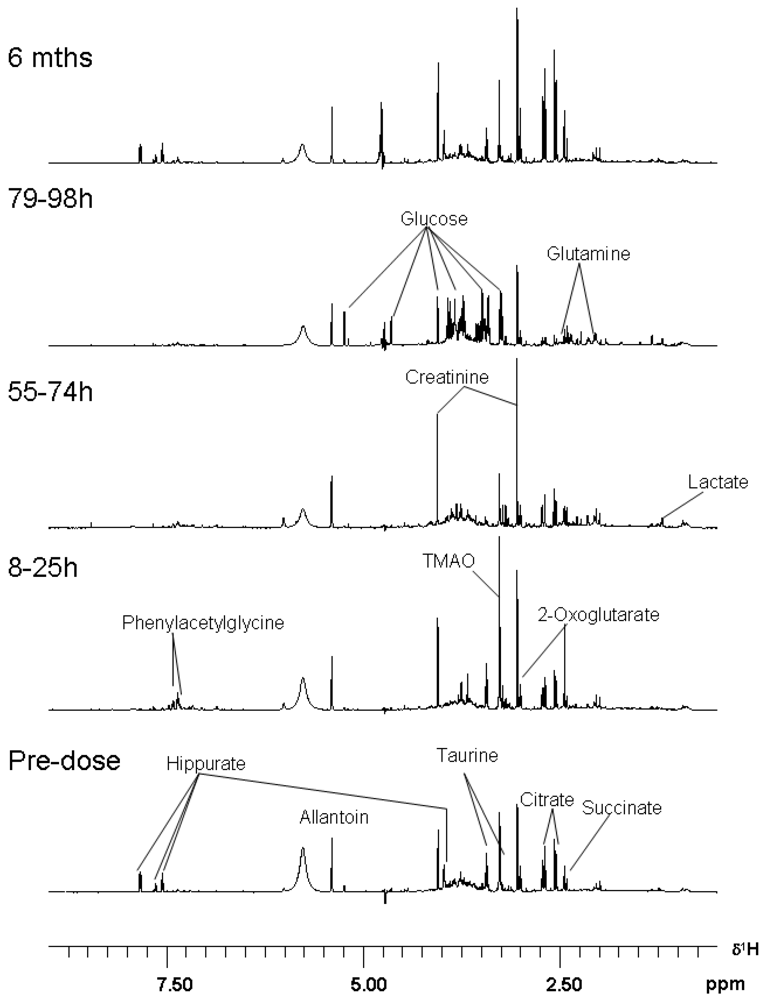
3.3. Metabolomics
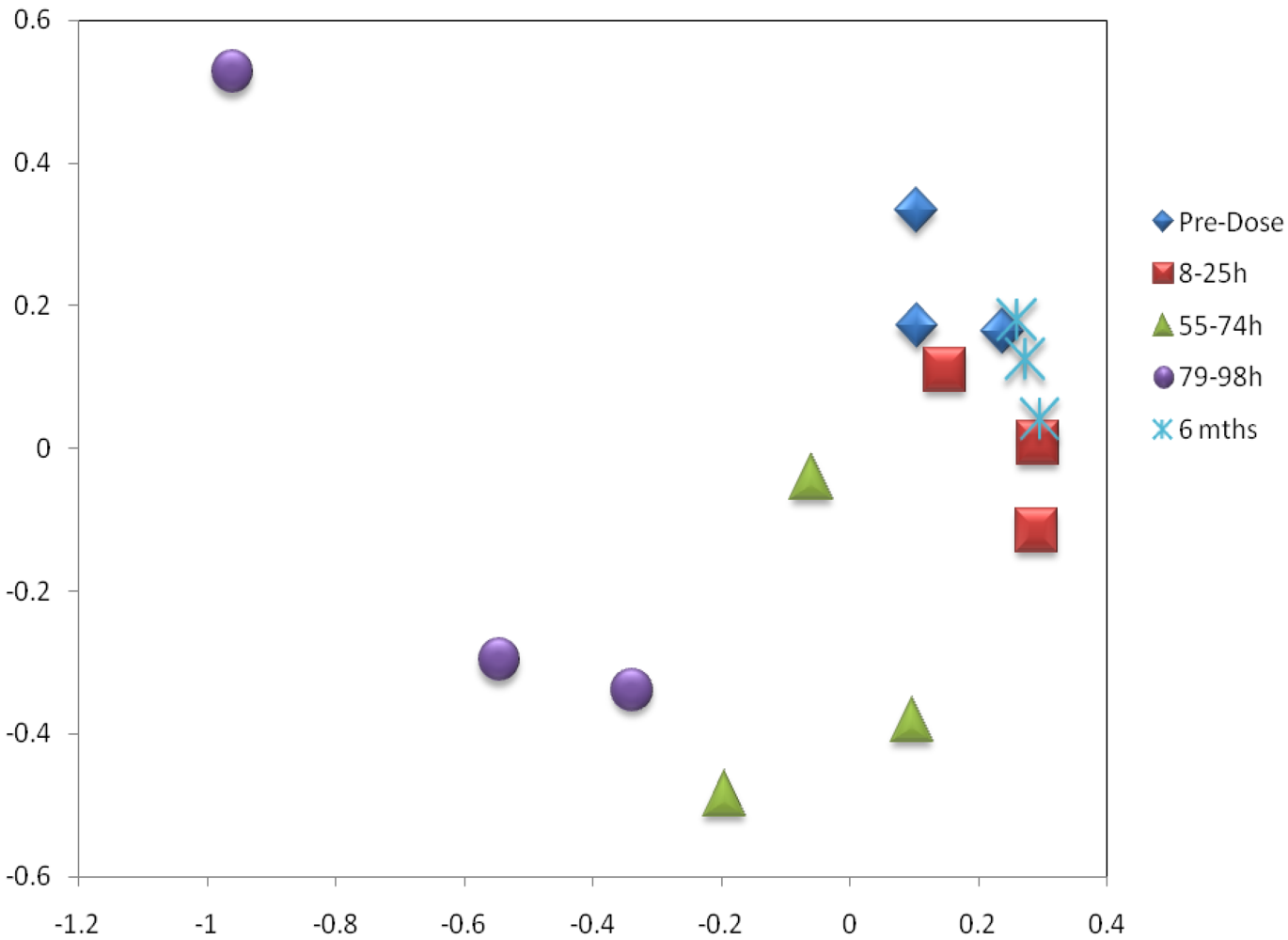
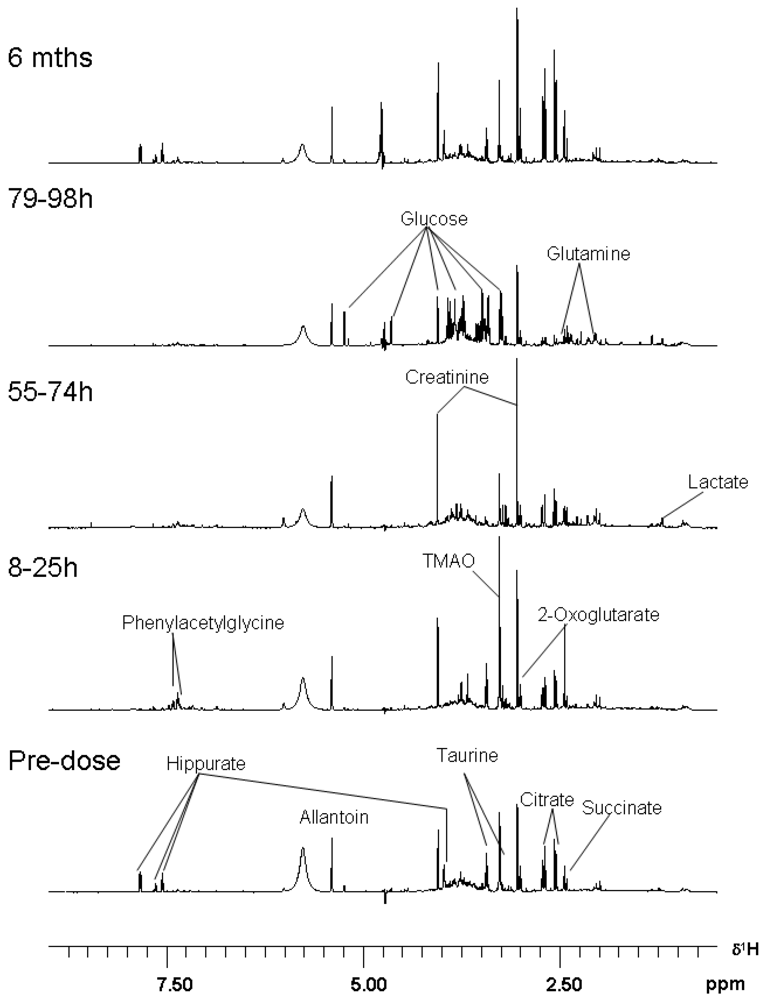
| Metabolite | Time-Period of Change | Delta |
|---|---|---|
| Hippurate | 8–25 | decrease |
| Phenylacetylglycine | 8–25 | increase |
| TMAO | 8–25 | increase |
| Citrate | 55–74 | increase |
| 2-Oxoglutarate | 55–74 | increase |
| Succinate | 55–74 | increase |
| Allantoin | 55–74 | increase |
| Creatinine | 55–74 | increase |
| Taurine | 55–74 | decrease |
| TMAO | 55–74 | decrease |
| Hippurate | 55–74 | decrease |
| Glucose | 79–98 h | increase |
| Glutamine | 79–98 h | increase |
| Lactate | 79–98 h | increase |
| Hippurate | 79–98 h | decrease |
| Citrate | 79–98 h | decrease |
4. Discussion
Acknowledgements
Conflict of Interest
- The authors declare no conflict of interest.
References
- JECFA. Ochratoxin A; World Health Organisation: Geneva, Switzerland, 2001; 47, pp. 1–132. Available online: http://www.inche-m.org/documents/jecfa/jecmono/v47je04.htm (accessed on 24 May 2011).
- EFSA. Opinion of the scientific panel on contaminants in the food chain on a request from the Commission related to ochratoxin A in food. EFSA J. 2006, 365, 1–56.
- Zepnik, H.; Volkel, W.; Dekant, W. Toxicokinetics of the mycotoxin ochratoxin A in F 344 rats after oral administration. Toxicol. Appl. Pharmacol. 2003, 192, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Mally, A.; Volkel, W.; Amberg, A.; Kurz, M.; Wanek, P.; Eder, E.; Hard, G.; Dekant, W. Functional, biochemical, and pathological effects of repeated oral administration of ochratoxin A to rats. Chem.Res. Toxicol. 2005, 18, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Sieber, M.; Wagner, S.; Rached, E.; Amberg, A.; Mally, A.; Dekant, W. Metabonomic study of ochratoxin A toxicity in rats after repeated administration: Phenotypic anchoring enhances the ability for biomarker discovery. Chem. Res. Toxicol. 2009, 22, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Boorman, G.A. Toxicology and Carcinogenesis Studies of Ochratoxin A. (CAS No. 303-47-9) in F344/N Rats, National Toxicology Program Technical Report; US Department of Health and Human Services, National Institutes of Health: Research Triangle Park, NC, USA, 1989; NIH Publication No. 89-2813. [Google Scholar]
- Waddell, W.J.; Fukushima, S.; Williams, G.M. Concordance of thresholds for carcinogenicity of N-nitrosodiethylamine. Arch. Toxicol. 2006, 80, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.; Kulinskaya, E.; Nestler, S. Renal tumourigenesis in male rats in response to chronic dietary ochratoxin A. Food Addit. Contam 2005, 22 (Suppl. 1), 58–64. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.; Kulinskaya, E. Lifetime, low-dose ochratoxin A, dietary study on renal carcinogenesis in male Fischer rats. Food Addit. Contam. 2010, 27, 1566–1573. [Google Scholar] [CrossRef]
- Mantle, P.G. Minimum tolerable exposure period and maximum threshold dietary intake of ochratoxin A for causing renal cancer in male Dark Agouti rats. Food Chem. Toxicol. 2009, 47, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.P.; Mantle, P.G. Biosynthesis of ochratoxins by Aspergillus ochraceus. Phytochemistry 2001, 58, 709–716. [Google Scholar] [PubMed]
- Purchase, I.F.C.; Theron, J.J. The acute toxicity of ochratoxin A to rats. Food Cosmet. Toxicol. 1968, 6, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Burtis, C.A.; Ashwood, E.R.; Bruns, D.E. Tietz Fundamentals of Clinical Chemistry; WB Saunders Company: Philadelphia, PA, USA, 2005. [Google Scholar]
- Stoev, S.D.; Paskalev, M.; Macdonald, S.; Mantle, P.G. Experimental one year ochratoxin A toxicosis in pigs. Exp. Toxicol. Pathol. 2002, 53, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.G. Interpretation of the pharmacokinetics of ochratoxin A in blood plasma of rats, during and after acute or chronic ingestion. Food Chem. Toxicol. 2008, 46, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Ensner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [PubMed]
- Mantle, P.G.; McHugh, K.M.; Adatia, R.; Gray, T.; Turner, D.R. Persistent karyomegaly caused by Penicillium nephrotoxins in the rat. Proc. R. Soc. Lond. Ser. B 1991, 246, 251–259. [Google Scholar] [CrossRef]
- Arbillaga, L.; Vettorazzi, A.; Gil, A.G.; van Delft, J.H.M.; Garcia-Jalon, J.A.; Lopez de Cerain, A. Gene expression changes induced by ochratoxin A in renal and hepatic tissues of male F344 rat after repeated oral administration. Toxicol. Appl. Pharmacol. 2008, 230, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.G.; Faucet-Marquis, V.; Manderville, R.A.; Squillaci, B.; Pfohl-Leszkowicz, A. Structures of covalent adducts between DNA and ochratoxin A: A new factor in debate about genotoxicity and human risk assessment. Chem. Res. Toxicol. 2010, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.G.; Nolan, C.C. Pathological outcomes in kidney and brain in male Fischer rats given dietary ochratoxin A, commencing at one year of age. Toxins 2010, 2, 1100–1110. [Google Scholar] [CrossRef]
- Waddell, W.J.; Fukushima, S.; Williams, G.M. Concordance of thresholds for carcinogenicity of N-nitrosodiethylamine. Arch. Toxicol. 2006, 80, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Storen, O.; Holm, H.; Stormer, F.C. Metabolism of ochratoxin A by rats. Appl. Environ. Microbiol. 1982, 44, 785–789. [Google Scholar] [PubMed]
- Vettorazzi, A.; Gonzalez-Peñas, E.; Troconiz, I.F.; Arbillaga, L.; Corcuera, L.A.; Gil, A.G.; Lopez de Cerain, A. A different kinetic profile of ochratoxin A in mature male rats. Food Chem. Toxicol. 2009, 47, 1921–1927. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, A.; Trocóniz, I.F.; Gonzalez-Peñas, E.; Corcuera, L.A.; Arbillaga, L.; Gil, A.G.; Nagy, J.M.; Mantle, P.G.; Lopez de Cerain, A. Effects of fasting and gender on ochratoxin A toxicokinetics in F344 rats. Food Chem. Toxicol. 2010, 48, 3159–3166. [Google Scholar]
- Beynon, R.J.; Hurst, J.L. Urinary proteins and the modulation of chemical scents in mice and rats. Peptides 2004, 25, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Treacy, E.P.; Akerman, B.R.; Chow, L.M.L.; Youil, R.; Bibeau, C.; Lin, J.; Bruce, A.G.; Knight, M.; Danks, D.M.; Cashman, J.R.; et al. Mutations of the flavin-containing monooxygenase gene (FMO3) cause trimethylaminuria, a defect in detoxication. Hum. Mol. Genet. 1998, 7, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Gartland, K.P.; Bonner, F.W.; Nicholson, J.K. Investigations into the biochemical effects of region-specific nephrotoxins. Mol. Pharmacol. 1989, 35, 242–250. [Google Scholar] [PubMed]
- Nicholls, A.W.; Mortishire-Smith, R.J.; Nicholson, J.K. NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatising germ-free rats. Chem. Res. Toxicol. 2003, 16, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Connor, S.C.; Wu, W.; Sweatman, B.C.; Manini, J.; Haselden, J.N.; Crowther, D.J.; Waterfield, C.J. Effects of feeding and body weight loss on the 1H NMR-based urine metabolic profiles of male Wistar Han rats: implications for biomarker discovery. Biomarkers 2004, 9, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Foxall, P.J.D.; Spraul, M.; Farrant, R.D.; Lindon, J.C. 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal. Chem. 1995, 67, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Holmes, E.; Nicholson, J.K.; Cloarec, O.; Chollet, J.; Tanner, M.; Singer, B.H.; Utzinger, J. Metabonomic investigations in mice infected with Schistosoma mansoni: An approach for biomarker identification. Proc. Natl. Acad. Sci. USA 2004, 101, 12676–12681. [Google Scholar]
- Li, S.; Marquardt, R.R.; Frohlich, A.A.; Vitti, T.G.; Crow, G. Pharmacokinetics of ochratoxin A and its metabolites in rats. Toxicol. Appl. Pharmacol. 1997, 145, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Jutabha, P.; Endou, H. Molecular mechanism of ochratoxin A transport in the kidney. Toxins 2010, 2, 1381–1398. [Google Scholar] [CrossRef]
- Garcia-Sanz, A.; Rodriguez-Barbero, A.; Bentley, M.D.; Ritman, E.L.; Romero, C.L. Three-dimensional microcomputed tomography of renal vasculature in rats. Hypertension 1998, 31, 440–451. [Google Scholar] [PubMed]
- Jennings-Gee, J.E.; Tozlovanu, M.; Manderville, R.; Miller, M.S.; Pfohl-Leszkowicz, A.; Schwartz, G.G. Ochratoxin A: In utero exposure in mice induces adducts in testicular DNA. Toxins 2010, 2, 1428–1444. [Google Scholar] [CrossRef] [PubMed]
Supplementary Files
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mantle, P.G.; Nicholls, A.W.; Shockcor, J.P. 1H NMR Spectroscopy-Based Metabolomic Assessment of Uremic Toxicity, with Toxicological Outcomes, in Male Rats Following an Acute, Mid-Life Insult from Ochratoxin A. Toxins 2011, 3, 504-519. https://doi.org/10.3390/toxins3060504
Mantle PG, Nicholls AW, Shockcor JP. 1H NMR Spectroscopy-Based Metabolomic Assessment of Uremic Toxicity, with Toxicological Outcomes, in Male Rats Following an Acute, Mid-Life Insult from Ochratoxin A. Toxins. 2011; 3(6):504-519. https://doi.org/10.3390/toxins3060504
Chicago/Turabian StyleMantle, Peter G., Andrew W. Nicholls, and John P. Shockcor. 2011. "1H NMR Spectroscopy-Based Metabolomic Assessment of Uremic Toxicity, with Toxicological Outcomes, in Male Rats Following an Acute, Mid-Life Insult from Ochratoxin A" Toxins 3, no. 6: 504-519. https://doi.org/10.3390/toxins3060504
APA StyleMantle, P. G., Nicholls, A. W., & Shockcor, J. P. (2011). 1H NMR Spectroscopy-Based Metabolomic Assessment of Uremic Toxicity, with Toxicological Outcomes, in Male Rats Following an Acute, Mid-Life Insult from Ochratoxin A. Toxins, 3(6), 504-519. https://doi.org/10.3390/toxins3060504