Alpha-Latrotoxin Rescues SNAP-25 from BoNT/A-Mediated Proteolysis in Embryonic Stem Cell-Derived Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Embryonic Stem Cell Culture and Neuronal Differentiation
2.3. Immunoblotting
2.4. Time-Lapse Confocal Microscopy
3. Results and Discussion
3.1. Results
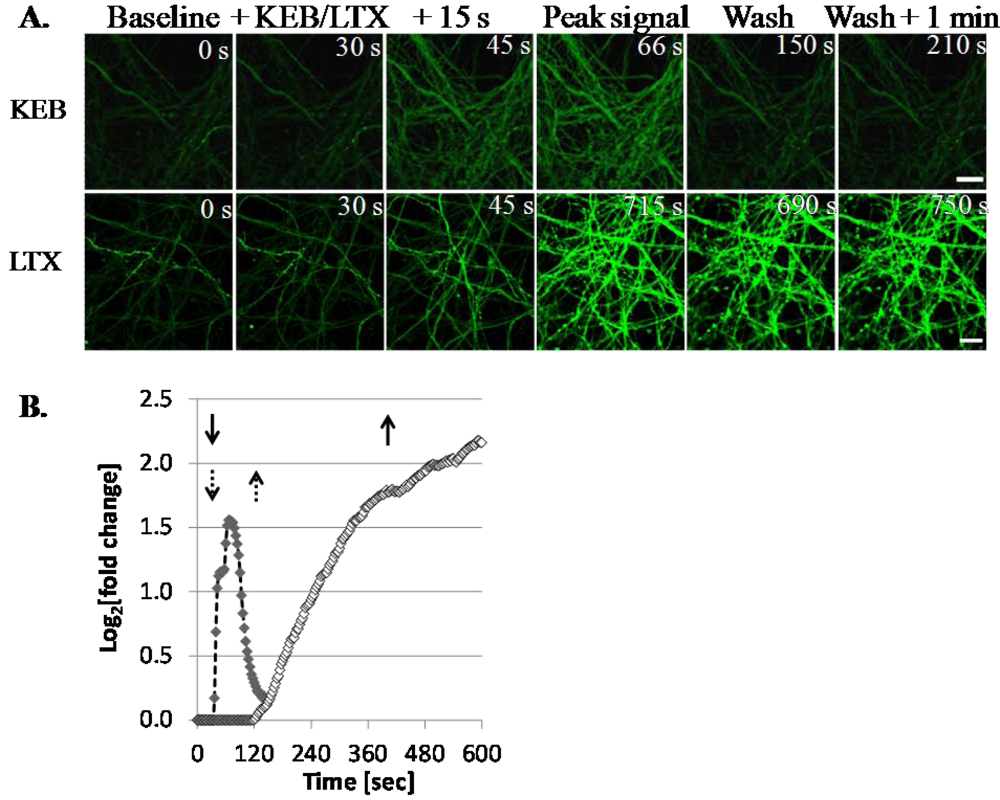
3.1.1. Optimization of the Screening Model
3.1.2. LTX Treatment of BoNT/A-Treated ESNs Restores Full-Length SNAP-25 Protein within 48 h.

3.1.3. LTX Treatment Results in Prolonged Ca2+ Internalization
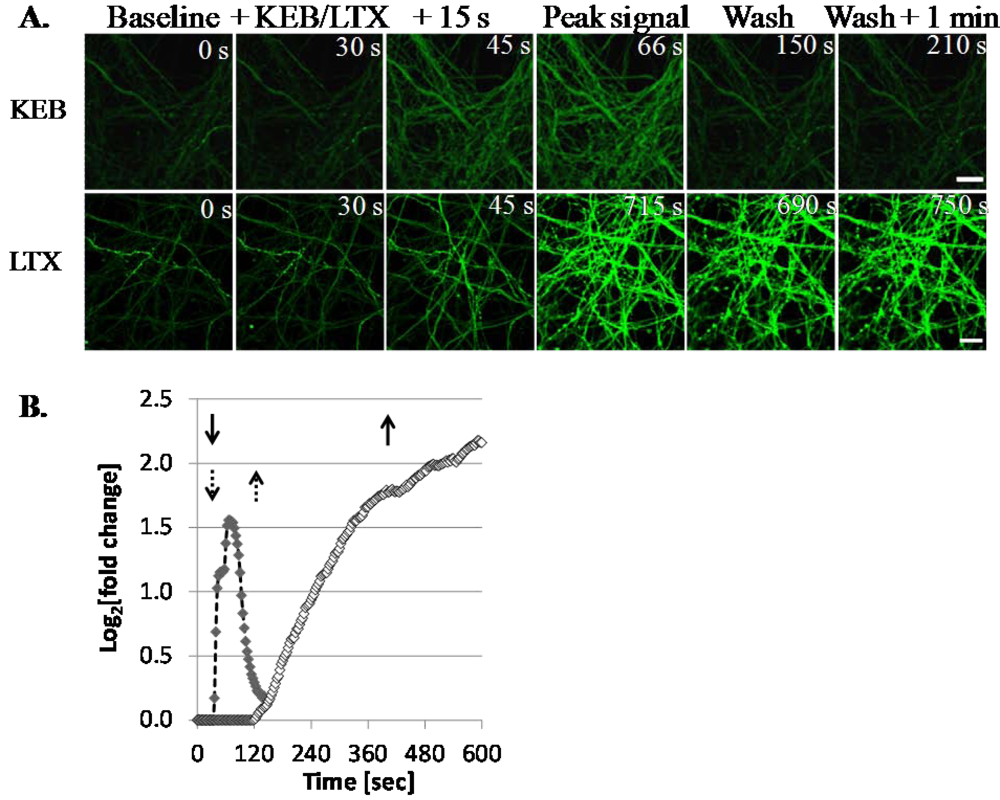
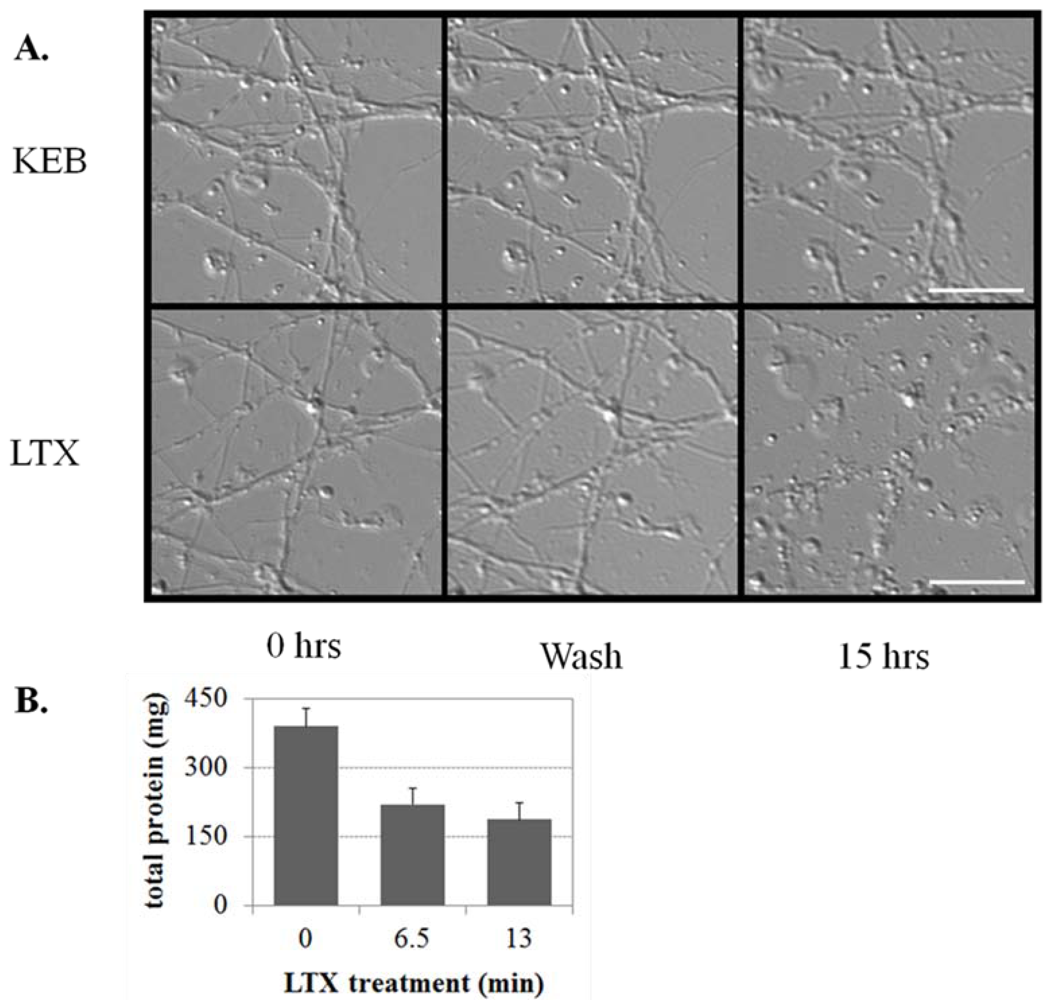
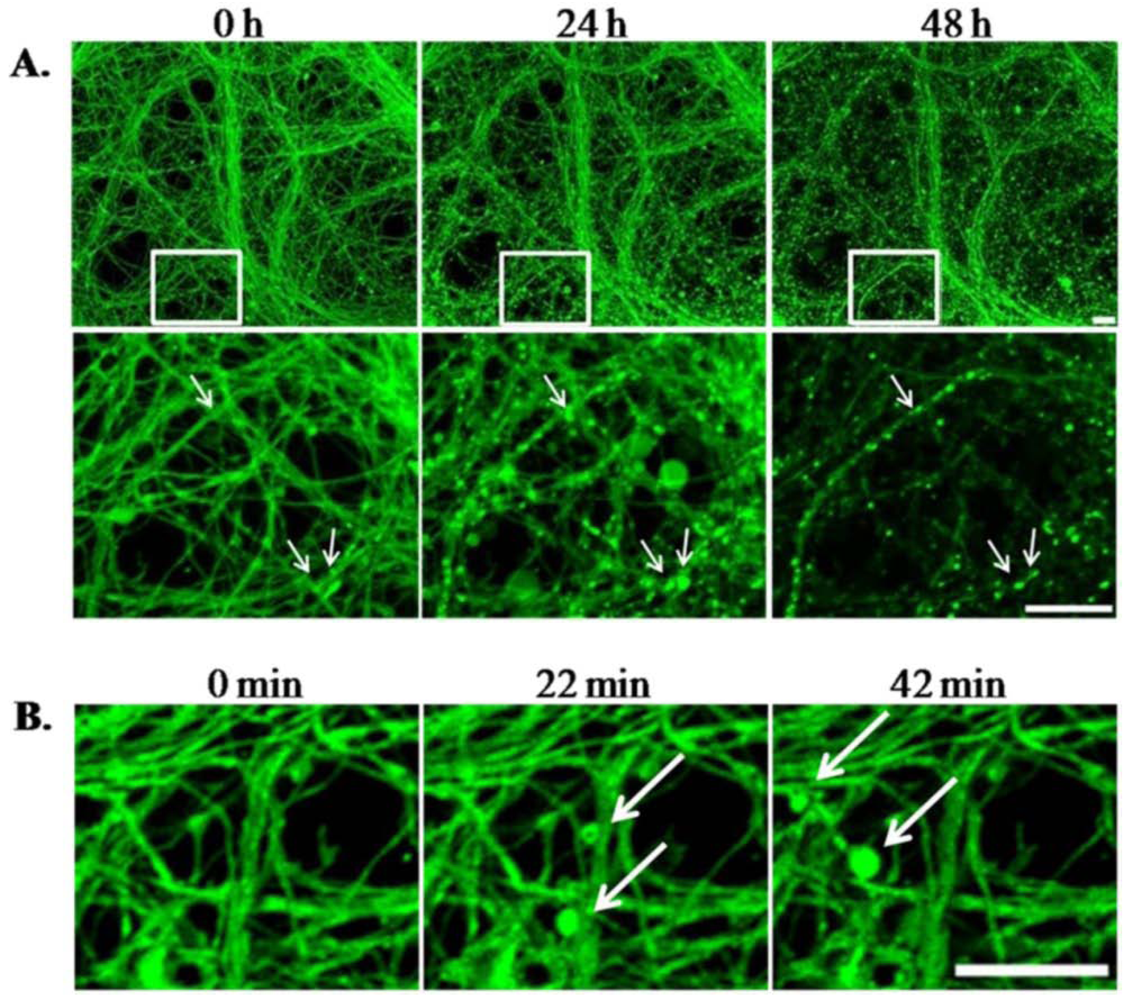
3.1.4. LTX-Treated ESNs Exhibit Evidence of Excitotoxicity That Partially Resolves between 24–48 h
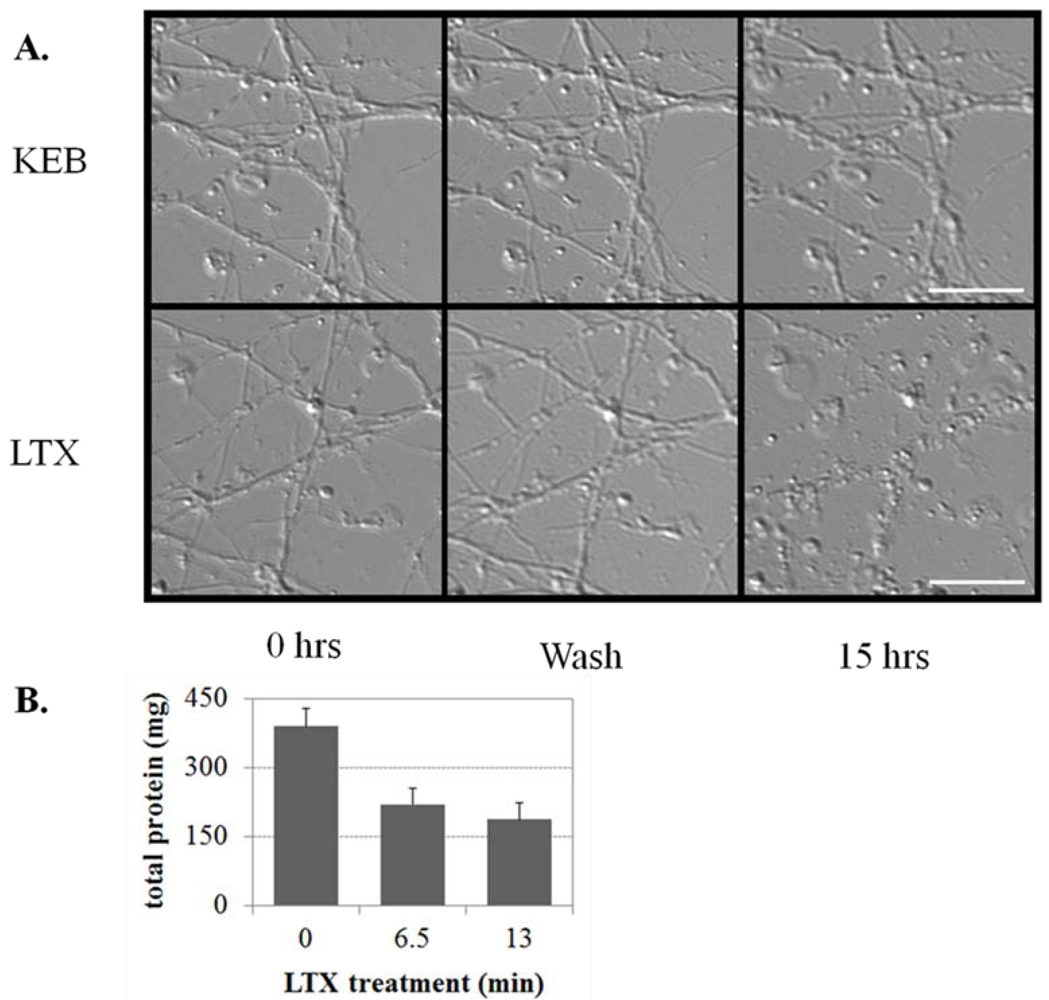

3.2. Discussion
3.2.1. Considerations in Developing a Screening Methodology
3.2.2. LTX Rescue of Full-Length SNAP-25 Expression
3.2.3. Is Neuronal Degeneration and/or Excitotoxicity Responsible for the Loss of BoNT Persistence?
3.2.4. ESNs Provide a Responsive, Genetically Tractable Model for LTX Research
4. Conclusions
Acknowledgements
References
- Simpson, L.L. Identification of the major steps in botulinum toxin action. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 167–193. [Google Scholar]
- Johnson, E.A.; Montecucco, C. Chapter 11 botulism. Handb. Clin. Neurol. 2008, 91, 333–368. [Google Scholar]
- Bakry, N.; Kamata, Y.; Simpson, L.L. Lectins from Triticum vulgaris and Limax flavus are universal antagonists of botulinum neurotoxin and tetanus toxin. J. Pharmacol. Exp. Ther. 1991, 258, 830–836. [Google Scholar]
- Adler, M.; Nicholson, J.D. Evaluation of toosendanin as a botulinum neurotoxin antagonist. Botulinum J. 2008, 1, 208–218. [Google Scholar]
- Larsen, J.C. U.S. Army Botulinum Neurotoxin (BoNT) Medical therapeutics research program: Past accomplishments and future directions. Drug Develop. Res. 2009, 70, 266–278. [Google Scholar] [CrossRef]
- Foran, P.G.; Mohammed, N.; Lisk, G.O.; Nagwaney, S.; Lawrence, G.W.; Johnson, E.; Smith, L.; Aoki, K.R.; Dolly, J.O. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 2003, 278, 1363–1371. [Google Scholar]
- Rogozhin, A.A.; Pang, K.K.; Bukharaeva, E.; Young, C.; Slater, C.R. Recovery of mouse neuromuscular junctions from single and repeated injections of botulinum neurotoxin A. J. Physiol. 2008, 586, 3163–3182. [Google Scholar]
- Morbiato, L.; Carli, L.; Johnson, E.A.; Montecucco, C.; Molgo, J.; Rossetto, O. Neuromuscular paralysis and recovery in mice injected with botulinum neurotoxins A and C. Eur. J. Neurosci. 2007, 25, 2697–2704. [Google Scholar]
- Juzans, P.; Comella, J.X.; Molgo, J.; Faille, L.; Angaut-Petit, D. Nerve terminal sprouting in botulinum type-A treated mouse levator auris longus muscle. Neuromuscul. Disord. 1996, 6, 177–185. [Google Scholar]
- Silva, J.P.; Suckling, J.; Ushkaryov, Y. Penelope’s web: Using alpha-latrotoxin to untangle the mysteries of exocytosis. J. Neurochem. 2009, 111, 275–290. [Google Scholar]
- Li, G.; Lee, D.; Wang, L.; Khvotchev, M.; Chiew, S.K.; Arunachalam, L.; Collins, T.; Feng, Z.P.; Sugita, S. N-terminal insertion and C-terminal ankyrin-like repeats of alpha-latrotoxin are critical for Ca2+-dependent exocytosis. J. Neurosci. 2005, 25, 10188–10197. [Google Scholar] [PubMed]
- Capogna, M.; Volynski, K.E.; Emptage, N.J.; Ushkaryov, Y.A. The alpha-latrotoxin mutant LTXN4C enhances spontaneous and evoked transmitter release in CA3 pyramidal neurons. J. Neurosci. 2003, 23, 4044–4053. [Google Scholar]
- Van Renterghem, C.; Iborra, C.; Martin-Moutot, N.; Lelianova, V.; Ushkaryov, Y.; Seagar, M. alpha-latrotoxin forms calcium-permeable membrane pores via interactions with latrophilin or neurexin. Eur. J. Neurosci. 2000, 12, 3953–3962. [Google Scholar]
- Ushkaryov, Y.A.; Rohou, A.; Sugita, S. alpha-Latrotoxin and its receptors. Handb. Exp. Pharmacol. 2008, 184, 171–206. [Google Scholar]
- Orlova, E.V.; Rahman, M.A.; Gowen, B.; Volynski, K.E.; Ashton, A.C.; Manser, C.; van Heel, M.; Ushkaryov, Y.A. Structure of alpha-latrotoxin oligomers reveals that divalent cation-dependent tetramers form membrane pores. Nat. Struct. Biol. 2000, 7, 48–53. [Google Scholar]
- Deak, F.; Liu, X.; Khvotchev, M.; Li, G.; Kavalali, E.T.; Sugita, S.; Südhof, T.C. Alpha-latrotoxin stimulates a novel pathway of Ca2+-dependent synaptic exocytosis independent of the classical synaptic fusion machinery. J. Neurosci. 2009, 29, 8639–8648. [Google Scholar]
- Bronk, P.; Deak, F.; Wilson, M.C.; Liu, X.; Sudhof, T.C.; Kavalali, E.T. Differential effects of SNAP-25 deletion on Ca2+ -dependent and Ca2+ -independent neurotransmission. J. Neurophysiol. 2007, 98, 794–806. [Google Scholar]
- Deak, F.; Schoch, S.; Liu, X.; Sudhof, T.C.; Kavalali, E.T. Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat. Cell. Biol. 2004, 6, 1102–1108. [Google Scholar]
- Washbourne, P.; Thompson, P.M.; Carta, M.; Costa, E.T.; Mathews, J.R.; Lopez-Benditó, G.; Molnár, Z.; Becher, M.W.; Valenzuela, C.F.; Partridge, L.D. et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat. Neurosci. 2002, 5, 19–26. [Google Scholar] [PubMed]
- Clark, A.W.; Hurlbut, W.P.; Mauro, A. Changes in the fine structure of the neuromuscular junction of the frog caused by black widow spider venom. J. Cell Biol. 1972, 52, 1–14. [Google Scholar]
- Ceccarelli, B.; Grohovaz, F.; Hurlbut, W.P. Freeze-fracture studies of frog neuromuscular junctions during intense release of neurotransmitter. II. Effects of electrical stimulation and high potassium. J. Cell. Biol. 1979, 81, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, B.; Grohovaz, F.; Hurlbut, W.P. Freeze-fracture studies of frog neuromuscular junctions during intense release of neurotransmitter. I. Effects of black widow spider venom and Ca2+-free solutions on the structure of the active zone. J. Cell. Biol. 1979, 81, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Duchen, L.W.; Gomez, S.; Queiroz, L.S. The neuromuscular junction of the mouse after black widow spider venom. J. Physiol. 1981, 316, 279–291. [Google Scholar]
- Henkel, A.W.; Betz, W.J. Monitoring of black widow spider venom (BWSV) induced exo- and endocytosis in living frog motor nerve terminals with FM1-43. Neuropharmacology 1995, 34, 1397–1406. [Google Scholar]
- Gomez, S.; Queiroz, L.S. The effects of black widow spider venom on the innervation of muscles paralysed by botulinum toxin. Q. J. Exp. Physiol. 1982, 67, 495–506. [Google Scholar]
- Thesleff, S.; Zelena, J.; Hofmann, W.W. Restoration of Function in Botulinum Paralysis by Experimental Nerve Regeneration. Proc. Soc. Exp. Biol. Med. 1964, 116, 19–20. [Google Scholar]
- Duchen, L.W. The effects in the mouse of nerve crush and regneration on the innervation of skeletal muscles paralysed by Clostridium botulinum toxin. J. Pathol. 1970, 102, 9–14. [Google Scholar]
- McNutt, P.; Celver, J.; Hamilton, T.; Mesngon, M. Embryonic stem cell-derived neurons are a novel, highly sensitive tissue culture platform for botulinum research. Biochem. Biophys. Res. Commun. 2011, 405, 85–90. [Google Scholar]
- Keller, J.E.; Neale, E.A.; Oyler, G.; Adler, M. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 1999, 456, 137–142. [Google Scholar]
- Sanders, J.D.; Yang, Y.; Liu, Y. Differential turnover of syntaxin and SNAP-25 during synaptogenesis in cultured cerebellar granule neurons. J. Neurosci. Res. 1998, 53, 670–676. [Google Scholar]
- Tedesco, E.; Rigoni, M.; Caccin, P.; Grishin, E.; Rossetto, O.; Montecucco, C. Calcium overload in nerve terminals of cultured neurons intoxicated by alpha-latrotoxin and snake PLA2 neurotoxins. Toxicon 2009, 54, 138–144. [Google Scholar]
- Boldt, G.E.; Eubanks, L.M.; Janda, K.D. Identification of a botulinum neurotoxin A protease inhibitor displaying efficacy in a cellular model. Chem. Commun. (Camb.) 2006, 7, 3063–3065. [Google Scholar]
- Dong, M.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Using fluorescent sensors to detect botulinum neurotoxin activity in vitro and in living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14701–14706. [Google Scholar]
- Puffer, E.B.; Lomneth, R.B.; Sarkar, H.K.; Singh, B.R. Differential roles of developmentally distinct SNAP-25 isoforms in the neurotransmitter release process. Biochemistry 2001, 40, 9374–9378. [Google Scholar]
- Purkiss, J.R.; Friis, L.M.; Doward, S.; Quinn, C.P. Clostridium botulinum neurotoxins act with a wide range of potencies on SH-SY5Y human neuroblastoma cells. Neurotoxicology 2001, 22, 447–453. [Google Scholar]
- Tarasenko, A.S.; Storchak, L.G.; Himmelreich, N.H. alpha-Latrotoxin affects mitochondrial potential and synaptic vesicle proton gradient of nerve terminals. Neurochem. Int. 2008, 52, 392–400. [Google Scholar]
- Ahmed, S.A.; Ludivico, M.L.; Smith, L.A. Factors affecting autocatalysis of botulinum A neurotoxin light chain. Protein J. 2004, 23, 445–451. [Google Scholar]
- de Paiva, A.; Meunier, F.A.; Molgo, J.; Aoki, K.R.; Dolly, J.O. Functional repair of motor endplates after botulinum neurotoxin type A poisoning: Biphasic switch of synaptic activity between nerve sprouts and their parent terminals. Proc. Natl. Acad. Sci. USA 1999, 96, 3200–3205. [Google Scholar]
- Chen, Q.; Olney, J.W.; Lukasiewicz, P.D.; Almli, T.; Romano, C. Ca2+-independent excitotoxic neurodegeneration in isolated retina, an intact neural net, a role for Cl- and inhibitory transmitters. Mol. Pharmacol. 1998, 53, 564–572. [Google Scholar] [PubMed]
- Hoffmann, E.K.; Dunham, P.B. Membrane mechanisms and intracellular signalling in cell volume regulation. Int. Rev.Cytol. 1995, 161, 173–262. [Google Scholar]
- Hasbani, M.J.; Hyrc, K.L.; Faddis, B.T.; Romano, C.; Goldberg, M.P. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp. Neurol. 1998, 154, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Ashton, A.C.; Volynski, K.E.; Lelianova, V.G.; Orlova, E.V.; van Renterghem, C.; Canepari, M.; Seagar, M.; Ushkaryov, Y.A. alpha-Latrotoxin, acting via two Ca2+-dependent pathways, triggers exocytosis of two pools of synaptic vesicles. J. Biol. Chem. 2001, 276, 44695–44703. [Google Scholar] [PubMed]
- Lajus, S.; Vacher, P.; Huber, D.; Dubois, M.; Benassy, M.N.; Ushkaryov, Y.; Lang, J. Alpha-latrotoxin induces exocytosis by inhibition of voltage-dependent K+ channels and by stimulation of L-type Ca2+ channels via latrophilin in beta-cells. J. Biol. Chem. 2006, 281, 5522–5531. [Google Scholar]
- Madl, J.E.; Burgesser, K. Adenosine triphosphate depletion reverses sodium-dependent, neuronal uptake of glutamate in rat hippocampal slices. J. Neurosci. 1993, 13, 4429–4444. [Google Scholar]
- Raiteri, L.; Stigliani, S.; Zedda, L.; Raiteri, M.; Bonanno, G. Multiple mechanisms of transmitter release evoked by “pathologically” elevated extracellular [K+]: Involvement of transporter reversal and mitochondrial calcium. J. Neurochem. 2002, 80, 706–714. [Google Scholar]
- Kubo, T.; Randolph, M.A.; Groger, A.; Winograd, J.M. Embryonic stem cell-derived motor neurons form neuromuscular junctions in vitro and enhance motor functional recovery in vivo. Plast. Reconstr. Surg. 2009, 123, 139S–148S. [Google Scholar] [CrossRef] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mesngon, M.; McNutt, P. Alpha-Latrotoxin Rescues SNAP-25 from BoNT/A-Mediated Proteolysis in Embryonic Stem Cell-Derived Neurons. Toxins 2011, 3, 489-503. https://doi.org/10.3390/toxins3050489
Mesngon M, McNutt P. Alpha-Latrotoxin Rescues SNAP-25 from BoNT/A-Mediated Proteolysis in Embryonic Stem Cell-Derived Neurons. Toxins. 2011; 3(5):489-503. https://doi.org/10.3390/toxins3050489
Chicago/Turabian StyleMesngon, Mariano, and Patrick McNutt. 2011. "Alpha-Latrotoxin Rescues SNAP-25 from BoNT/A-Mediated Proteolysis in Embryonic Stem Cell-Derived Neurons" Toxins 3, no. 5: 489-503. https://doi.org/10.3390/toxins3050489
APA StyleMesngon, M., & McNutt, P. (2011). Alpha-Latrotoxin Rescues SNAP-25 from BoNT/A-Mediated Proteolysis in Embryonic Stem Cell-Derived Neurons. Toxins, 3(5), 489-503. https://doi.org/10.3390/toxins3050489