Consequences and Utility of the Zinc-Dependent Metalloprotease Activity of Anthrax Lethal Toxin

Abstract
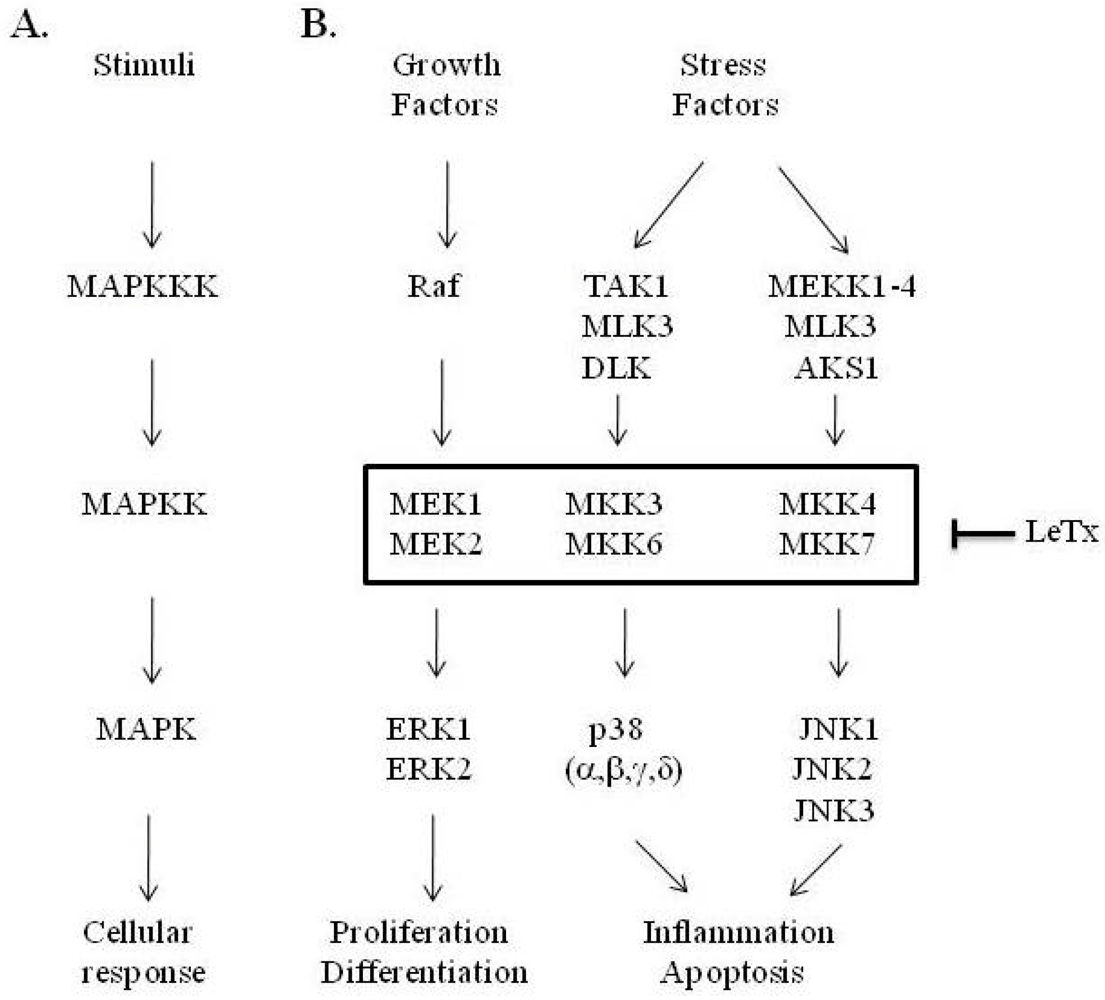
:1. Introduction
2. Structure and Function of Lethal Factor
2.1. Identification of functional domains of LF
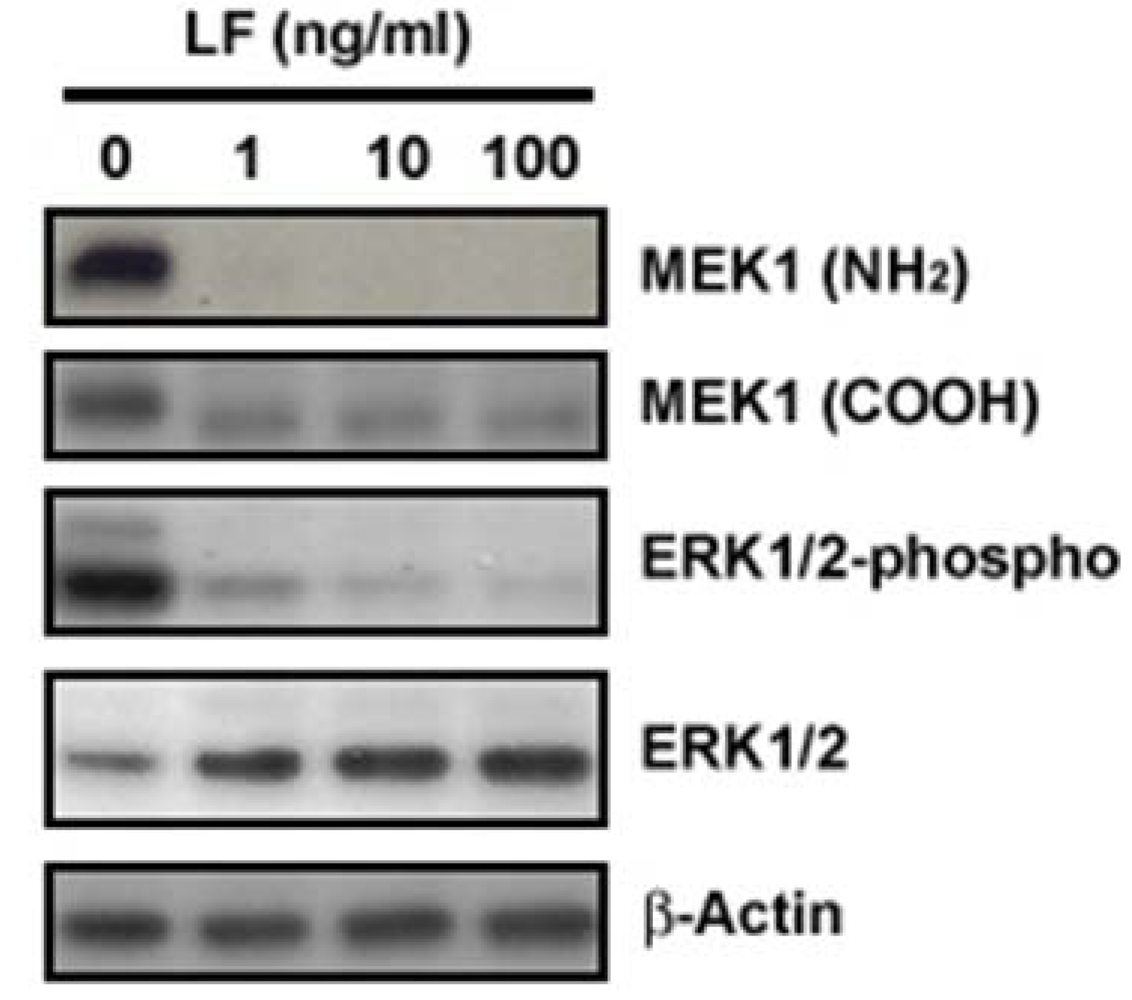
2.2. LF is a zinc-dependent metalloprotease
3. LF Zinc-Metalloprotease Activity Is Specific for the MAPK Pathway
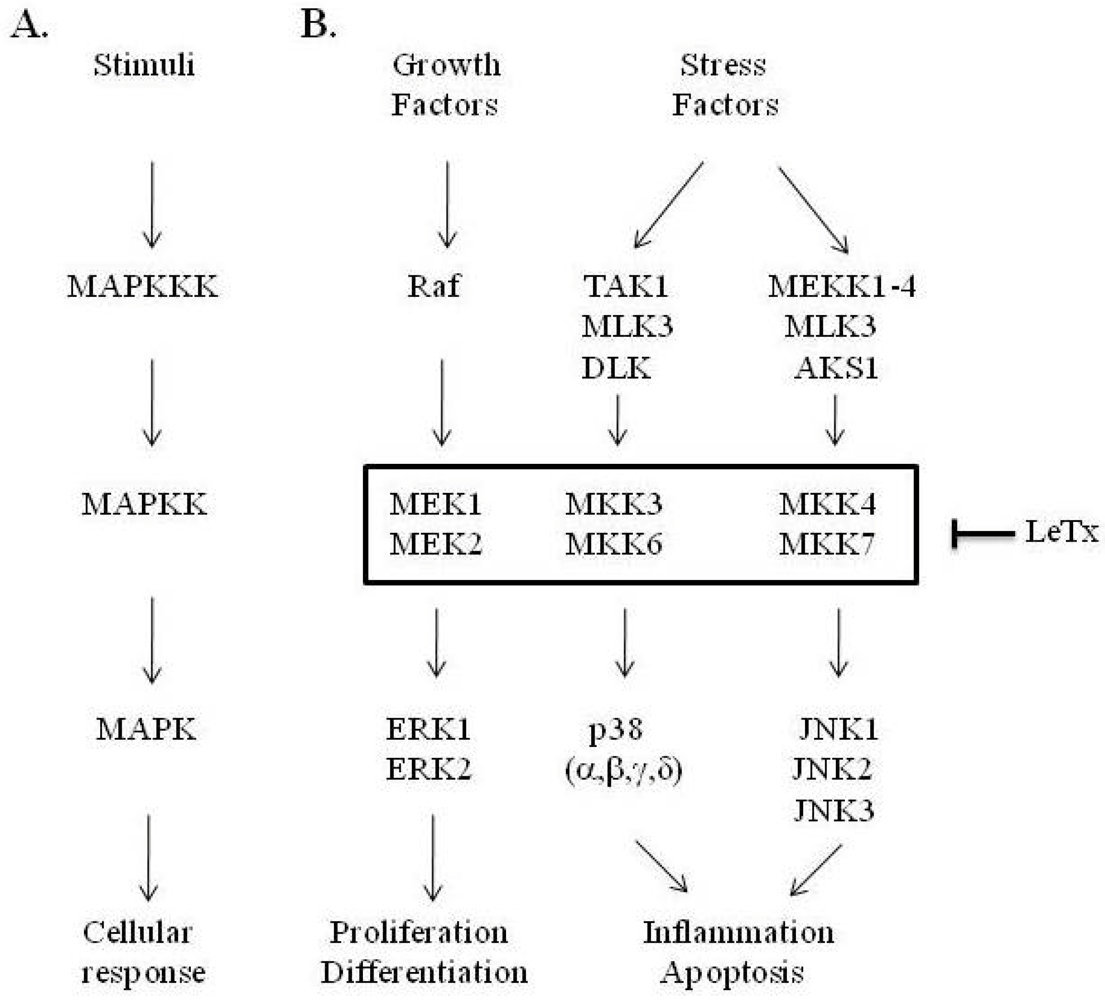
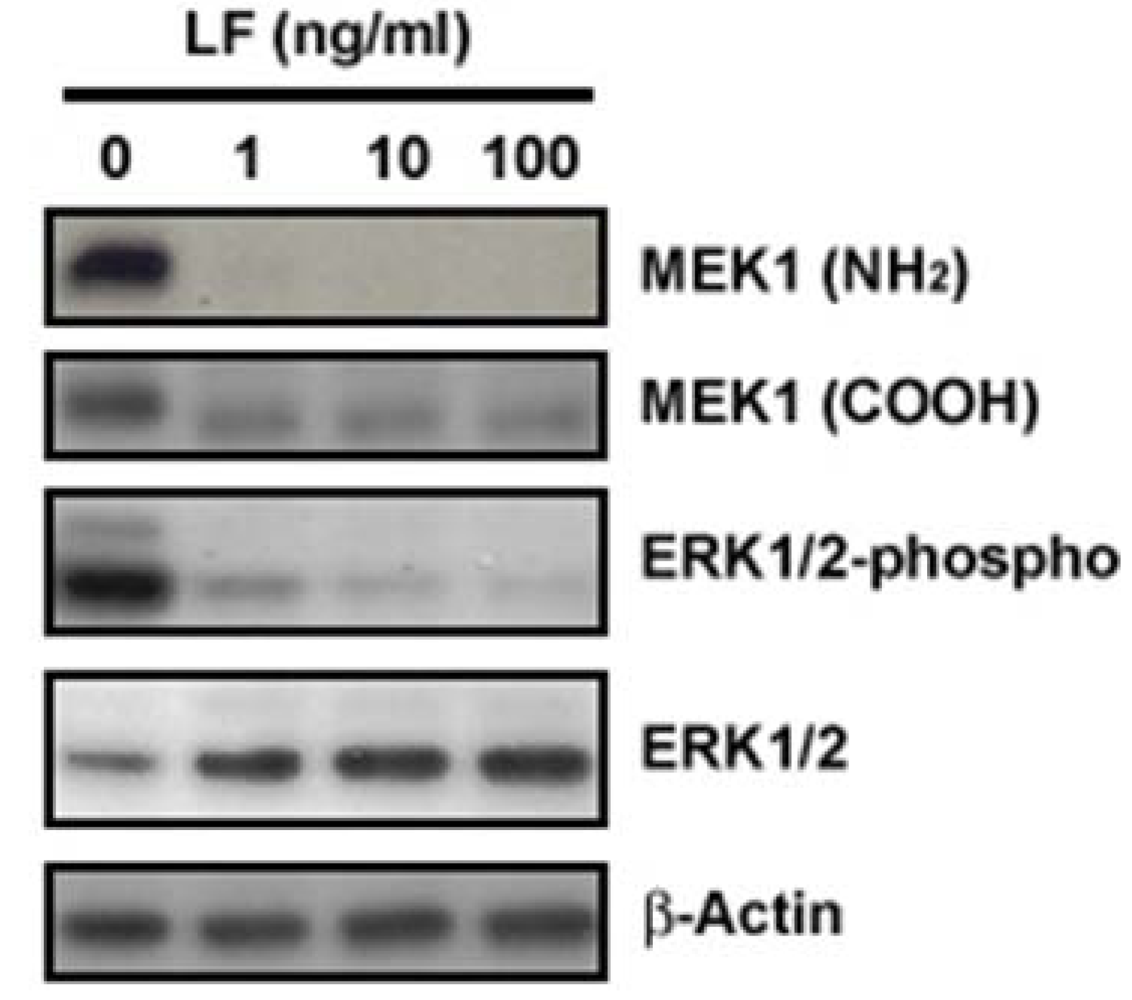

{kind=link}
{kind=link}
{kind=link}
| LF substrates | Amino acid sequences | |
|---|---|---|
| MEK1 | M PKKK P T P(8) | / I Q L N P A P D |
| MEK2 | A RRKP V L P(11) | / A L T I N P T I |
| MKK3 | S KRKK D L R(26) | / I S C M S K P P |
| MKK4(K45-L46) | Q G KRK A L K(45) | / L N F A N P P F |
| MKK4(R58-F59) | P P F K S T A R(58) | / F T L N P N P T |
| MKK6 | K KR N P G L K(14) | / I P K E A F E Q |
| MKK7(Q44-L45) | Q RPRP T L Q(44) | / L P L A N D G G |
| MKK7(Q76-L77) | A RPRH M L G(76) | / L P S T L F T P |
| Consensus sequence | (B/P)3-4 X X X | / Al |
4. Consequences of the Zinc-Dependent Metalloprotease Activity of LF
4.1. Immune modulation
4.2. Vascular damage
5. The Utility of the MEK-Dependent Metalloprotease Activity of LF in Other Pathological Conditions
5.1. Tumor growth and angiogenesis
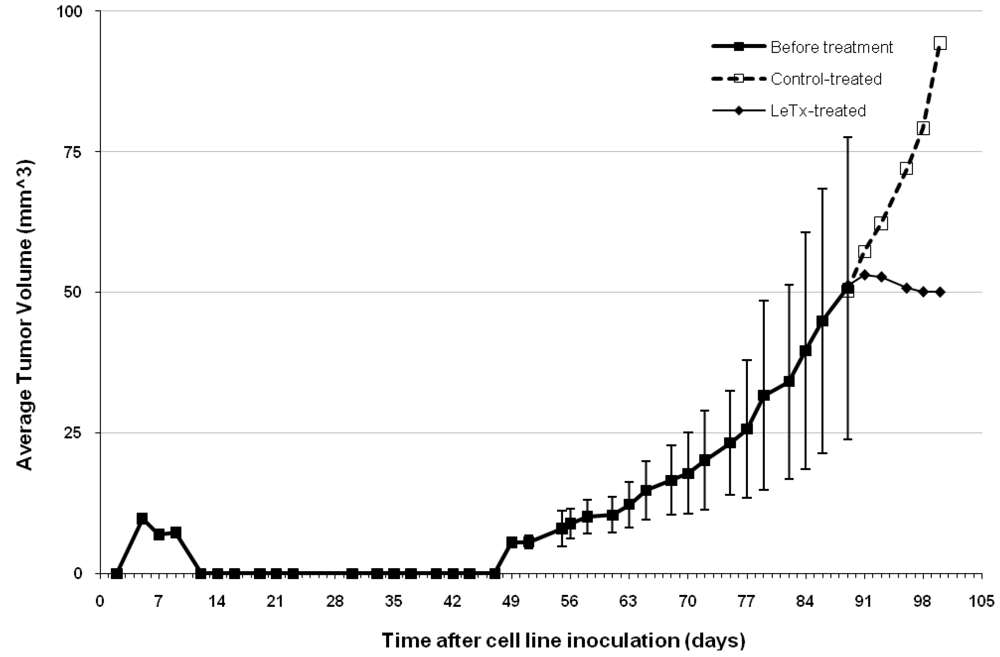
5.2. Retinal Neovascularization
6. Conclusions
| Effect | Anthrax [16,21,24,97] | Tumor [32,38,64,68] | Retina [84] |
|---|---|---|---|
| Macrophage activity | Altered phagocytosis, Septic shock-like syndrome | n.d. | n.d. |
| Cytokine release | mixed effects in vitro and in vivo | Largely depressed in vitro and in vivo | Largely depressed in vitro and in vivo sequelae-elevated VEGF |
| Vascular function | Hemorrhage, vascular permeability, hypoxia, hypotension | Decreased perfusion, hemorrhage, decreased mean vessel density | Decreased perfusion, block in branching morphogenesis |
| Consequence | Hypotensive shock, death | Decreased tumor volume | Retinopathy |
Acknowledgements
References and Notes
- Smith, H.; Keppie, J. bservations on experimental anthrax; demonstration of a specific lethal factor produced in vivo by Bacillus anthracis. Nature 1954, 173, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Vande Woude, G.F. Anthrax toxins. Cell. Mol. Life Sci. 1999, 55, 1599–1609. [Google Scholar] [CrossRef]
- Singh, Y.; Leppla, S.H.; Bhatnagar, R.; Friedlander, A.M. Internalization and processing of Bacillus anthracis lethal toxin by toxin-sensitive and -resistant cells. J. Biol. Chem. 1989, 264, 11099–11102. [Google Scholar]
- Stanley, J.L.; Smith, H. Purification of factor I and recognition of a third factor of the anthrax toxin. J. Gen. Microbiol. 1961, 26, 49–63. [Google Scholar]
- Beall, F.A.; Taylor, M.J.; Thorne, C.B. Rapid lethal effect in rats of a third component found upon fractionating the toxin of Bacillus anthracis. J. Bacteriol. 1962, 83, 1274–1280. [Google Scholar]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar]
- Moayeri, M.; Wiggins, J.F.; Leppla, S.H. Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect. Immun. 2007, 75, 5175–5184. [Google Scholar] [CrossRef]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar] [PubMed]
- O'Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar]
- Voth, D.E.; Hamm, E.E.; Nguyen, L.G.; Tucker, A.E.; Salles, II; Ortiz-Leduc, W.; Ballard, J.D. Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell Microbiol. 2005, 7. [Google Scholar]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H. The roles of anthrax toxin in pathogenesis. Curr. Opin. Microbiol. 2004, 7, 19–24. [Google Scholar] [CrossRef]
- Sherer, K.; Li, Y.; Cui, X.; Eichacker, P.Q. Lethal and edema toxins in the pathogenesis of Bacillus anthracis septic shock: Implications for therapy. Am. J. Respir Crit. Care Med. 2007, 175, 211–221. [Google Scholar]
- Tournier, J.N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects Med. 2009, 30, 456–466. [Google Scholar] [CrossRef]
- Friedlander, A.M.; Bhatnagar, R.; Leppla, S.H.; Johnson, L.; Singh, Y. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 1993, 61, 245–52. [Google Scholar]
- Agrawal, A.; Lingappa, J.; Leppla, S.H.; Agrawal, S.; Jabbar, A.; Quinn, C.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003, 424, 329–334. [Google Scholar]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D'Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef]
- Guarner, J.; Jernigan, J.A.; Shieh, W.J.; Tatti, K.; Flannagan, L.M.; Stephens, D.S.; Popovic, T.; Ashford, D.A.; Perkins, B.A.; Zaki, S.R. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 2003, 163, 701–709. [Google Scholar] [CrossRef]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar] [CrossRef]
- Warfel, J.M.; Steele, A.D.; D'Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar] [CrossRef]
- Kuo, S.R.; Willingham, M.C.; Bour, S.H.; Andreas, E.A.; Park, S.K.; Jackson, C.; Duesbery, N.S.; Leppla, S.H.; Tang, W.J.; Frankel, A.E. Anthrax toxin-induced shock in rats is associated with pulmonary edema and hemorrhage. Microb. Pathog. 2008, 44, 467–472. [Google Scholar] [CrossRef]
- Bolcome, R.E., III; Sullivan, S.E.; Zeller, R.; Barker, A.P.; Collier, R.J.; Chan, J. Anthrax lethal toxin induces cell death-independent permeability in zebrafish vasculature. Proc. Natl. Acad. Sci. USA 2008, 105, 2439–2444. [Google Scholar]
- Mikesell, P.; Ivins, B.E.; Ristroph, J.D.; Dreier, T.M. Evidence for plasmid-mediated toxin production in Bacillus anthracis. Infect. Immun. 1983, 39, 371–376. [Google Scholar]
- Tonello, F.; Montecucco, C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 2009, 30, 431–438. [Google Scholar] [CrossRef]
- Singh, Y.; Liang, X.; Duesbery, N.S. Pathogenesis of Bacillus anthracis: The Role of Anthrax Toxins. In Microbial Toxins: Molecular and Cellular Biology; Proft, T., Ed.; Horizon Bioscience: Norfolk, UK, 2005; pp. 285–312. [Google Scholar]
- Quinn, C.P.; Singh, Y.; Klimpel, K.R.; Leppla, S.H. Functional mapping of anthrax toxin lethal factor by in-frame insertion mutagenesis. J. Biol. Chem. 1991, 266, 20124–20130. [Google Scholar]
- Klimpel, K.R.; Arora, N.; Leppla, S.H. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol. Microbiol. 1994, 13, 1093–1100. [Google Scholar]
- Kochi, S.K.; Schiavo, G.; Mock, M.; Montecucco, C. Zinc content of the Bacillus anthracis lethal factor. FEMS Microbiol. Lett. 1994, 124, 343–348. [Google Scholar] [CrossRef]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; Vande Woude, G.F. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar] [CrossRef]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 Pt 3, 739–745. [Google Scholar]
- Duesbery, N.S.; Resau, J.; Webb, C.P.; Koochekpour, S.; Koo, H.M.; Leppla, S.H.; Vande Woude, G.F. Suppression of ras-mediated transformation and inhibition of tumor growth and angiogenesis by anthrax lethal factor, a proteolytic inhibitor of multiple MEK pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 4089–4094. [Google Scholar]
- Tanoue, T.; Adachi, M.; Moriguchi, T.; Nishida, E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell. Biol. 2000, 2, 110–116. [Google Scholar] [CrossRef]
- Bardwell, A.J.; Flatauer, L.J.; Matsukuma, K.; Thorner, J.; Bardwell, L. A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 2001, 276, 10374–10386. [Google Scholar]
- Chopra, A.P.; Boone, S.A.; Liang, X.; Duesbery, N.S. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 2003, 278, 9402–9406. [Google Scholar]
- Bardwell, A.J.; Abdollahi, M.; Bardwell, L. Anthrax lethal factor-cleavage products of MAPK (mitogen-activated protein kinase) kinases exhibit reduced binding to their cognate MAPKs. Biochem. J. 2004, 378, 569–577. [Google Scholar] [CrossRef]
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar]
- Banks, D.J.; Ward, S.C.; Bradley, K.A. New insights into the functions of anthrax toxin. Expert Rev. Mol. Med. 2006, 8, 1–18. [Google Scholar]
- Rossi Paccani, S.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C.T. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell Microbiol. 2007, 9, 924–929. [Google Scholar] [CrossRef]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and p38 MAPK in the immune system: Signal integration, propagation and termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef]
- Buscher, D.; Hipskind, R.A.; Krautwald, S.; Reimann, T.; Baccarini, M. Ras-dependent and -independent pathways target the mitogen-activated protein kinase network in macrophages. Mol. Cell Biol. 1995, 15, 466–475. [Google Scholar]
- Kugler, S.; Schuller, S.; Goebel, W. Involvement of MAP-kinases and -phosphatases in uptake and intracellular replication of Listeria monocytogenes in J774 macrophage cells. FEMS Microbiol. Lett. 1997, 157, 131–136. [Google Scholar] [CrossRef]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar] [CrossRef] [PubMed]
- Pages, G.; Guerin, S.; Grall, D.; Bonino, F.; Smith, A.; Anjuere, F.; Auberger, P.; Pouyssegur, J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999, 286, 1374–1377. [Google Scholar]
- Yamashita, M.; Kimura, M.; Kubo, M.; Shimizu, C.; Tada, T.; Perlmutter, R.M.; Nakayama, T. T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 1024–1029. [Google Scholar]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef]
- Rincon, M.; Davis, R.J. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 2009, 228, 212–224. [Google Scholar] [CrossRef]
- Rincon, M.; Enslen, H.; Raingeaud, J.; Recht, M.; Zapton, T.; Su, M.S.; Penix, L.A.; Davis, R.J.; Flavell, R.A. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998, 17, 2817–2829. [Google Scholar]
- Dong, C.; Yang, D.D.; Wysk, M.; Whitmarsh, A.J.; Davis, R.J.; Flavell, R.A. Defective T cell differentiation in the absence of Jnk1. Science 1998, 282, 2092–2095. [Google Scholar]
- Lu, H.T.; Yang, D.D.; Wysk, M.; Gatti, E.; Mellman, I.; Davis, R.J.; Flavell, R.A. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 1999, 18, 1845–1857. [Google Scholar] [CrossRef]
- Iijima, N.; Yanagawa, Y.; Clingan, J.M.; Onoe, K. CCR7-mediated c-Jun N-terminal kinase activation regulates cell migration in mature dendritic cells. Int. Immunol. 2005, 17, 1201–1212. [Google Scholar] [CrossRef]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Duesbery, N.S.; Frankel, A.E. Potent inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin: Implications for broad anti-tumor efficacy. Cell Cycle 2008, 7, 745–9. [Google Scholar] [CrossRef]
- Depeille, P.E.; Ding, Y.; Bromberg-White, J.L.; Duesbery, N.S. MKK signaling and vascularization. Oncogene 2007, 26, 1290–1296. [Google Scholar] [CrossRef]
- Giroux, S.; Tremblay, M.; Bernard, D.; Cardin-Girard, J.F.; Aubry, S.; Larouche, L.; Rousseau, S.; Huot, J.; Landry, J.; Jeannotte, L.; Charron, J. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 1999, 9, 369–372. [Google Scholar]
- Hatano, N.; Mori, Y.; Oh-hora, M.; Kosugi, A.; Fujikawa, T.; Nakai, N.; Niwa, H.; Miyazaki, J.; Hamaoka, T.; Ogata, M. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 2003, 8, 847–856. [Google Scholar] [CrossRef]
- Wang, X.; Merritt, A.J.; Seyfried, J.; Guo, C.; Papadakis, E.S.; Finegan, K.G.; Kayahara, M.; Dixon, J.; Boot-Handford, R.P.; Cartwright, E.J.; Mayer, U.; Tournier, C. Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol. Cell Biol. 2005, 25, 336–345. [Google Scholar]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar]
- Koo, H.M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Woude, G.F. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar]
- Reddy, K.B.; Nabha, S.M.; Atanaskova, N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003, 22, 395–403. [Google Scholar] [CrossRef]
- Abi-Habib, R.J.; Singh, R.; Leppla, S.H.; Greene, J.J.; Ding, Y.; Berghuis, B.; Duesbery, N.S.; Frankel, A.E. Systemic anthrax lethal toxin therapy produces regressions of subcutaneous human melanoma tumors in athymic nude mice. Clin. Cancer Res. 2006, 12, 7437–7443. [Google Scholar]
- Depeille, P.; Young, J.J.; Boguslawski, E.A.; Berghuis, B.D.; Kort, E.J.; Resau, J.H.; Frankel, A.E.; Duesbery, N.S. Anthrax lethal toxin inhibits growth of and vascular endothelial growth factor release from endothelial cells expressing the human herpes virus 8 viral G protein coupled receptor. Clin. Cancer Res. 2007, 13, 5926–5934. [Google Scholar]
- Ding, Y.; Boguslawski, E.A.; Berghuis, B.D.; Young, J.J.; Zhang, Z.; Hardy, K.; Furge, K.; Kort, E.; Frankel, A.E.; Hay, R.V.; Resau, J.H.; Duesbery, N.S. Mitogen-activated protein kinase kinase signaling promotes growth and vascularization of fibrosarcoma. Mol. Cancer Ther. 2008, 7, 648–658. [Google Scholar]
- Ugurel, S.; Rappl, G.; Tilgen, W.; Reinhold, U. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J. Clin. Oncol. 2001, 19, 577–583. [Google Scholar]
- Rogers, M.S.; Christensen, K.A.; Birsner, A.E.; Short, S.M.; Wigelsworth, D.J.; Collier, R.J.; D'Amato, R.J. Mutant anthrax toxin B moiety (protective antigen) inhibits angiogenesis and tumor growth. Cancer Res. 2007, 67, 9980–9985. [Google Scholar]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef]
- Hotchkiss, K.A.; Basile, C.M.; Spring, S.C.; Bonuccelli, G.; Lisanti, M.P.; Terman, B.I. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp. Cell Res. 2005, 305, 133–144. [Google Scholar] [CrossRef]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar] [PubMed]
- Munoz-Chapuli, R.; Quesada, A.R.; Angel Medina, M. Angiogenesis and signal transduction in endothelial cells. Cell. Mol. Life Sci. 2004, 61, 2224–2243. [Google Scholar]
- Pages, G.; Berra, E.; Milanini, J.; Levy, A.P.; Pouyssegur, J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J. Biol. Chem. 2000, 275, 26484–26491. [Google Scholar]
- Milanini, J.; Vinals, F.; Pouyssegur, J.; Pages, G. p42/p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J. Biol. Chem. 1998, 273, 18165–18172. [Google Scholar]
- Wu, L.W.; Mayo, L.D.; Dunbar, J.D.; Kessler, K.M.; Baerwald, M.R.; Jaffe, E.A.; Wang, D.; Warren, R.S.; Donner, D.B. Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J. Biol. Chem. 2000, 275, 5096–5103. [Google Scholar]
- D'Angelo, G.; Struman, I.; Martial, J.; Weiner, R.I. Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc. Natl. Acad. Sci. USA 1995, 92, 6374–6378. [Google Scholar]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef]
- Hood, J.D.; Bednarski, M.; Frausto, R.; Guccione, S.; Reisfeld, R.A.; Xiang, R.; Cheresh, D.A. Tumor regression by targeted gene delivery to the neovasculature. Science 2002, 296, 2404–2407. [Google Scholar]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; Cao, Y.; Shujath, J.; Gawlak, S.; Eveleigh, D.; Rowley, B.; Liu, L.; Adnane, L.; Lynch, M.; Auclair, D.; Taylor, I.; Gedrich, R.; Voznesensky, A.; Riedl, B.; Post, L.E.; Bollag, G.; Trail, P.A. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar]
- Mavria, G.; Vercoulen, Y.; Yeo, M.; Paterson, H.; Karasarides, M.; Marais, R.; Bird, D.; Marshall, C.J. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 2006, 9, 33–44. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Currie, B.M.; Molinolo, A.; Leung, H.J.; Moayeri, M.; Basile, J.R.; Alfano, R.W.; Gutkind, J.S.; Frankel, A.E.; Bugge, T.H.; Leppla, S.H. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J. Biol. Chem. 2008, 283, 529–540. [Google Scholar]
- Bromberg-White, J.L.; Boguslawski, E.; Duesbery, N.S. Perturbation of mouse retinal vascular morphogenesis by anthrax lethal toxin. PLoS One 2009, 4, e6956. [Google Scholar]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar]
- Saint-Geniez, M.; D'Amore, P.A. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int. J. Dev. Biol. 2004, 48, 1045–1058. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Hackett, S.F. Ocular neovascularization: a valuable model system. Oncogene 2003, 22, 6537–6548. [Google Scholar] [CrossRef]
- Kvanta, A. Ocular angiogenesis: The role of growth factors. Acta Ophthalmol. Scand. 2006, 84, 282–288. [Google Scholar] [CrossRef]
- Kulkarni, A.D.; Kuppermann, B.D. Wet age-related macular degeneration. Adv. Drug. Deliv. Rev. 2005, 57, 1994–2009. [Google Scholar] [CrossRef]
- Hecquet, C.; Lefevre, G.; Valtink, M.; Engelmann, K.; Mascarelli, F. Activation and role of MAP kinase-dependent pathways in retinal pigment epithelial cells: ERK and RPE cell proliferation. Invest. Ophthalmol. Vis. Sci. 2002, 43, 3091–3098. [Google Scholar]
- Hecquet, C.; Lefevre, G.; Valtink, M.; Engelmann, K.; Mascarelli, F. Activation and role of MAP kinase-dependent pathways in retinal pigment epithelium cells: JNK1, P38 kinase, and cell death. Invest. Ophthalmol. Vis. Sci. 2003, 44, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Bullard, L.E.; Qi, X.; Penn, J.S. Role for extracellular signal-responsive kinase-1 and -2 in retinal angiogenesis. Invest. Ophthalmol. Vis. Sci. 2003, 44, 1722–1731. [Google Scholar]
- Hayashi, A.; Koroma, B.M.; Imai, K.; de Juan, E., Jr. Increase of protein tyrosine phosphorylation in rat retina after ischemia-reperfusion injury. Invest Ophthalmol. Vis. Sci. 1996, 37, 2146–2156. [Google Scholar]
- Hayashi, A.; Imai, K.; Kim, H.C.; de Juan, E., Jr. Activation of protein tyrosine phosphorylation after retinal branch vein occlusion in cats. Invest. Ophthalmol. Vis. Sci. 1997, 38, 372–380. [Google Scholar]
- Guma, M.; Rius, J.; Duong-Polk, K.X.; Haddad, G.G.; Lindsey, J.D.; Karin, M. Genetic and pharmacological inhibition of JNK ameliorates hypoxia-induced retinopathy through interference with VEGF expression. Proc. Natl. Acad. Sci. USA 2009, 106, 8760–8765. [Google Scholar]
- Bromberg-White, J.L.; Boguslawski, E.; Duesbery, N.S. 2010. Van Andel Institute, Grand Rapids, MI. Unpublished work..
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003, 112, 670–682. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bromberg-White, J.; Lee, C.-S.; Duesbery, N. Consequences and Utility of the Zinc-Dependent Metalloprotease Activity of Anthrax Lethal Toxin. Toxins 2010, 2, 1038-1053. https://doi.org/10.3390/toxins2051038
Bromberg-White J, Lee C-S, Duesbery N. Consequences and Utility of the Zinc-Dependent Metalloprotease Activity of Anthrax Lethal Toxin. Toxins. 2010; 2(5):1038-1053. https://doi.org/10.3390/toxins2051038
Chicago/Turabian StyleBromberg-White, Jennifer, Chih-Shia Lee, and Nicholas Duesbery. 2010. "Consequences and Utility of the Zinc-Dependent Metalloprotease Activity of Anthrax Lethal Toxin" Toxins 2, no. 5: 1038-1053. https://doi.org/10.3390/toxins2051038
APA StyleBromberg-White, J., Lee, C.-S., & Duesbery, N. (2010). Consequences and Utility of the Zinc-Dependent Metalloprotease Activity of Anthrax Lethal Toxin. Toxins, 2(5), 1038-1053. https://doi.org/10.3390/toxins2051038