Spider-Venom Peptides as Therapeutics


 ,
, 
Abstract
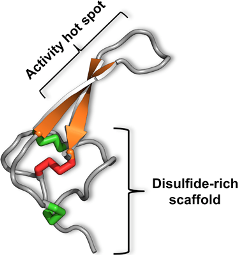
1. Introduction: The Diverse Pharmacology of Spider Venoms
2. Peptide Nomenclature
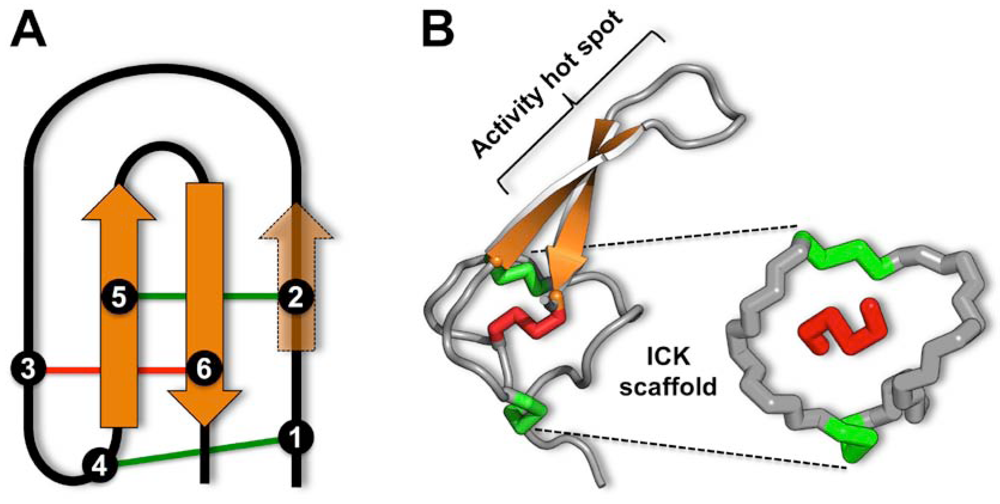
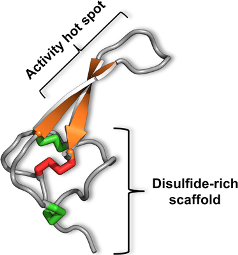
3. The Magical Properties of the Inhibitor Cystine Knot
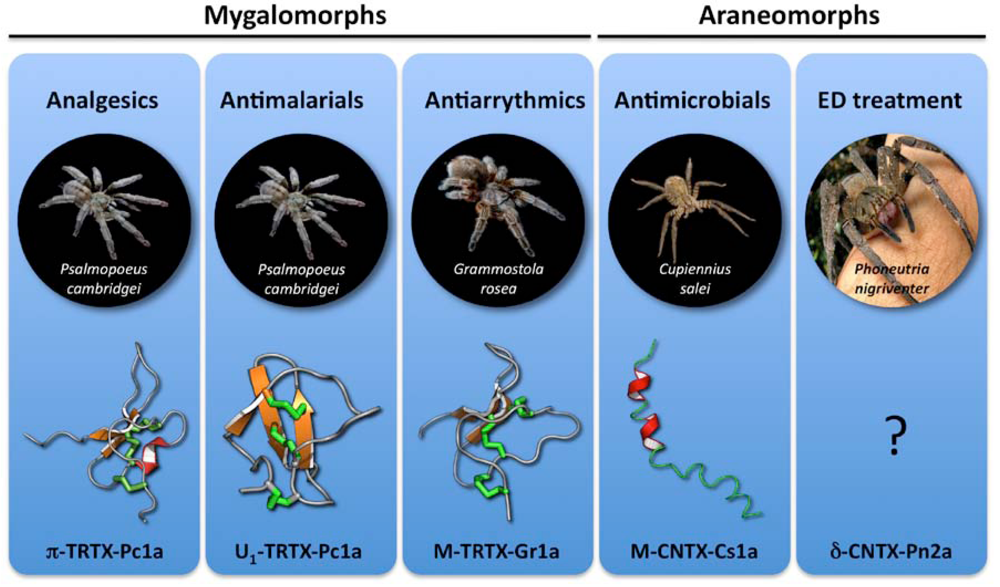
4. No Pain, Much Gain: Spider Toxins with Analgesic Potential
4.1. Modulators of Acid Sensing Ion Channels
4.2. Modulators of Voltage-Gated Sodium Channels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Name | No. of Residues | ICK Scaffold | IC50 (nM) against Various NaV Subtypes | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1.1 | 1.2 | 1.3 | 1.4 | 1.5 | 1.6 | 1.7 | 1.8 | |||
| β-TRTX-Tp1a | 35 | Yes | NA2 | NA | NA | NA | NA | NA | 51 | 273 |
| β-TRTX-Tp2a | 30 | Yes | NA | 41 | 102 | NA | 79 | 26 | 0.3 | 146 |
| β-TRTX-Ps1a | 34 | Yes | 610 | 0.6 | 42 | 288 | 72 | NA | NA | >1000 |
| β-TRTX-Cm1a | 33 | Yes | 523 | 3 | NA | 888 | 323 | NA | NA | >1000 |
| β-TRTX-Cm1b | 33 | Yes | 407 | 8 | 88 | 400 | 1634 | NA | NA | >2000 |
| δ-TRTX-Cj1a4 | 33 | Yes | NA | NA | NA | NA | 32 | 130 | 130 | NA |
4.3. Modulators of P2X Receptors
4.4 Spider-Venom Peptides that Modulate Other Pain Targets
5. Antiarrhythmic Drugs from Spider Venoms
6. Spider Toxins for Treating Erectile Dysfunction
7. Antibacterial and Antifungal Toxins
8. Antimalarial Toxins
9. Discussion
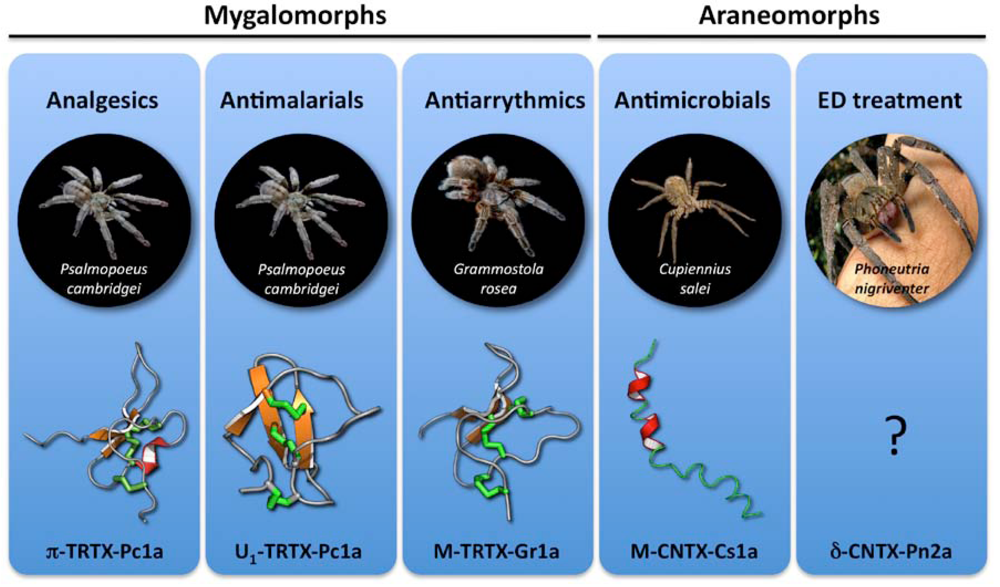
10. Conclusions
Acknowledgements
References
- Coddington, J.A.; Levi, H.W. Systematics and evolution of spiders (Araneae). Annu. Rev. Ecol. Syst. 1991, 22, 565–592. [Google Scholar]
- Isbister, G.K.; White, J. Clinical consequences of spider bite: recent advances in our understanding. Toxicon 2004, 43, 477–492. [Google Scholar]
- King, G.F. The wonderful world of spiders: preface to the special Toxicon issue on spider venoms. Toxicon 2004, 43, 471–476. [Google Scholar]
- Escoubas, P.; Diochot, S.; Corzo, G. Structure and pharmacology of spider venom neurotoxins. Biochimie 2000, 82, 893–907. [Google Scholar]
- Rash, L.D.; Hodgson, W.C. Pharmacology and biochemistry of spider venoms. Toxicon 2002, 40, 225–254. [Google Scholar]
- Tedford, H.W.; Sollod, B.L.; Maggio, F.; King, G.F. Australian funnel-web spiders: Master insecticide chemists. Toxicon 2004, 43, 601–618. [Google Scholar]
- Escoubas, P.; Rash, L. Tarantulas: Eight-legged pharmacists and combinatorial chemists. Toxicon 2004, 43, 555–574. [Google Scholar]
- Estrada, G.; Villegas, E.; Corzo, G. Spider venoms: a rich source of acylpolyamines and peptides as new leads for CNS drugs. Nat. Prod. Rep. 2007, 24, 145–161. [Google Scholar]
- Vassilevski, A.A.; Kozlov, S.A.; Grishin, E.V. Molecular diversity of spider venom. Biochemistry 2009, 74, 1505–1534. [Google Scholar]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar]
- Escoubas, P.; King, G.F. Venomics as a drug discovery platform. Expert Rev. Proteomics 2009, 6, 221–224. [Google Scholar]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar]
- Liao, Z.; Cao, J.; Li, S.; Yan, X.; Hu, W.; He, Q.; Chen, J.; Tang, J.; Xie, J.; Liang, S. Proteomic and peptidomic analysis of the venom from Chinese tarantula Chilobrachys jingzhao. Proteomics 2007, 7, 1892–1907. [Google Scholar]
- Richardson, M.; Pimenta, A.M.; Bemquerer, M.P.; Santoro, M.M.; Beirao, P.S.; Lima, M.E.; Figueiredo, S.G.; Bloch, C., Jr.; Vasconcelos, E.A.; Campos, F.A.; Gomes, P.C.; Cordeiro, M.N. Comparison of the partial proteomes of the venoms of Brazilian spiders of the genus Phoneutria. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2006, 142, 173–187. [Google Scholar] [PubMed]
- de F Fernandes-Pedrosa, M.; de LM Junqueira-de-Azevedo, I.; Gonçalves-de-Andrade, R.M.; Kobashi, L.S.; Almeida, D.D.; Ho, P.L.; Tambourgi, D.V. Transcriptome analysis of Loxosceles laeta (Araneae, Sicariidae) spider venomous gland using expressed sequence tags. BMC Genomics 2008, 9, 279. [Google Scholar] [PubMed]
- Jiang, L.; Peng, L.; Chen, J.; Zhang, Y.; Xiong, X.; Liang, S. Molecular diversification based on analysis of expressed sequence tags from the venom glands of the Chinese bird spider Ornithoctonus huwena. Toxicon 2008, 51, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.; Malyavka, A.; McCutchen, B.; Lu, A.; Schepers, E.; Herrmann, R.; Grishin, E. A novel strategy for the identification of toxinlike structures in spider venom. Proteins 2005, 59, 131–140. [Google Scholar]
- Diego-Garcia, E.; Peigneur, S.; Waelkens, E.; Debaveye, S.; Tytgat, J. Venom components from Citharischius crawshayi spider (Family Theraphosidae): Exploring transcriptome, venomics, and function. Cell. Mol. Life Sci. 2010, 67, 2799–2813. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Y.; Hu, W.; Xu, D.; Tao, H.; Yang, X.; Li, Y.; Jiang, L.; Liang, S. Molecular diversification of peptide toxins from the tarantula Haplopelma hainanum (Ornithoctonus hainana) venom based on transcriptomic, peptidomic, and genomic analyses. J. Proteome Res. 2010, 9, 2550–2564. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.L.; Miljenovic, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. ArachnoServer: A database of protein toxins from spiders. BMC Genomics 2009, 10, 375. [Google Scholar]
- Herzig, V.; Wood, D.L.A.; Newell, F.; Chaumeil, P.-A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. ArachnoServer 2.0, an updated online resource for spider toxin sequences and structures. Nucl. Acids Res. 2010. [Google Scholar]
- Gao, L.; Shan, B.E.; Chen, J.; Liu, J.H.; Song, D.X.; Zhu, B.C. Effects of spider Macrothele raven venom on cell proliferation and cytotoxicity in HeLa cells. Acta Pharmacol. Sin. 2005, 26, 369–376. [Google Scholar] [PubMed]
- Gao, L.; Yu, S.; Wu, Y.; Shan, B. Effect of spider venom on cell apoptosis and necrosis rates in MCF-7 cells. DNA Cell Biol. 2007, 26, 485–489. [Google Scholar]
- Ushkaryov, Y.A.; Volynski, K.E.; Ashton, A.C. The multiple actions of black widow spider toxins and their selective use in neurosecretion studies. Toxicon 2004, 43, 527–542. [Google Scholar]
- Swartz, K.J.; MacKinnon, R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 1995, 15, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.E. Agatoxins: Ion channel specific toxins from the American funnel web spider, Agelenopsis aperta. Toxicon 2004, 43, 509–525. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Modulation of insect CaV channels by peptidic spider toxins. Toxicon 2007, 49, 513–530. [Google Scholar]
- King, G.F.; Escoubas, P.; Nicholson, G.M. Peptide toxins that selectively target insect NaV and CaV channels. Channels 2008, 2, 100–116. [Google Scholar]
- Grishin, E.V.; Savchenko, G.A.; Vassilevski, A.A.; Korolkova, Y.V.; Boychuk, Y.A.; Viatchenko-Karpinski, V.Y.; Nadezhdin, K.D.; Arseniev, A.S.; Pluzhnikov, K.A.; Kulyk, V.B.; Voitenko, N.V.; Krishtal, O.O. Novel peptide from spider venom inhibits P2X3 receptors and inflammatory pain. Ann. Neurol. 2010, 67, 680–683. [Google Scholar]
- Escoubas, P.; De Weille, J.R.; Lecoq, A.; Diochot, S.; Waldmann, R.; Champigny, G.; Moinier, D.; Menez, A.; Lazdunski, M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 2000, 275, 25116–25121. [Google Scholar]
- Bowman, C.L.; Gottlieb, P.A.; Suchyna, T.M.; Murphy, Y.K.; Sachs, F. Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: history, properties, mechanisms and pharmacology. Toxicon 2007, 49, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar]
- Escoubas, P.; Bosmans, F. Spider peptide toxins as leads for drug development. Expert Opin. Drug Discov. 2007, 2, 823–835. [Google Scholar]
- Walker, M.J.A.; Barrett, T.; Guppy, L.J. Functional pharmacology: The drug discovery bottleneck? Drug Discov. Today 2004, 3, 208–215. [Google Scholar]
- Billingsley, M.L. Druggable targets and targeted drugs: enhancing the development of new therapeutics. Pharmacology 2008, 82, 239–244. [Google Scholar]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: a new paradigm for natural products-based drug discovery. Amino Acids 2010. [Google Scholar]
- Nestor, J.J., Jr. The medicinal chemistry of peptides. Curr. Med. Chem. 2009, 16, 4399–4418. [Google Scholar]
- Hamman, J.H.; Enslin, G.M.; Kotze, A.F. Oral delivery of peptide drugs: Barriers and developments. BioDrugs 2005, 19, 165–177. [Google Scholar]
- Pallaghy, P.K.; Nielsen, K.J.; Craik, D.J.; Norton, R.S. A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994, 3, 1833–1839. [Google Scholar]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar]
- King, G.F.; Tedford, H.W.; Maggio, F. Structure and function of insecticidal neurotoxins from Australian funnel-web spiders. J. Toxicol. Toxin Rev. 2002, 21, 359–389. [Google Scholar]
- Fletcher, J.I.; Chapman, B.E.; Mackay, J.P.; Howden, M.E.H.; King, G.F. The structure of versutoxin (δ-atracotoxin-Hv1) provides insights into the binding of site 3 neurotoxins to the voltage-gated sodium channel. Structure 1997, 5, 1525–1535. [Google Scholar]
- Fletcher, J.I.; Smith, R.; O'Donoghue, S.I.; Nilges, M.; Connor, M.; Howden, M.E.H.; Christie, M.J.; King, G.F. The structure of a novel insecticidal neurotoxin, ω-atracotoxin-HV1, from the venom of an Australian funnel web spider. Nat. Struct. Biol. 1997, 4, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.J.; Abbott, U.R.; Hatzos, C. Digestibility of food allergens and nonallergenic proteins in simulated gastric fluid and simulated intestinal fluid—A comparative study. J. Agric. Food Chem. 2002, 50, 7154–7160. [Google Scholar]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. 2010, 49, 6545–6548. [Google Scholar]
- Sollod, B.L.; Wilson, D.; Zhaxybayeva, O.; Gogarten, J.P.; Drinkwater, R.; King, G.F. Were arachnids the first to use combinatorial peptide libraries? Peptides 2005, 26, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.; Carr, D.B.; Cousins, M. Pain management: A fundamental human right. Anesth. Analg. 2007, 105, 205–221. [Google Scholar]
- The global pain market, 2008–2023; Visiongain ltd.: London, UK, 2008.
- Kellenberger, S.; Schild, L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol. Rev. 2002, 82, 735–767. [Google Scholar]
- Lingueglia, E. Acid-sensing ion channels in sensory perception. J. Biol. Chem. 2007, 282, 17325–17329. [Google Scholar]
- Gründer, S.; Chen, X. Structure, function, and pharmacology of acid-sensing ion channels (ASICs): Focus on ASIC1a. Int. J. Physiol. Pathophysiol. Pharmacol. 2010, 2, 73–94. [Google Scholar] [PubMed]
- Wemmie, J.A.; Price, M.P.; Welsh, M.J. Acid-sensing ion channels: Advances, questions and therapeutic opportunities. Trends Neurosci. 2006, 29, 578–586. [Google Scholar]
- Xiong, Z.G.; Pignataro, G.; Li, M.; Chang, S.Y.; Simon, R.P. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr. Opin. Pharmacol. 2008, 8, 25–32. [Google Scholar]
- Sluka, K.A.; Winter, O.C.; Wemmie, J.A. Acid-sensing ion channels: a new target for pain and CNS diseases. Curr. Opin. Drug Discov. Devel. 2009, 12, 693–704. [Google Scholar]
- Mazzuca, M.; Heurteaux, C.; Alloui, A.; Diochot, S.; Baron, A.; Voilley, N.; Blondeau, N.; Escoubas, P.; Gelot, A.; Cupo, A.; Zimmer, A.; Zimmer, A.M.; Eschalier, A.; Lazdunski, M. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci. 2007, 10, 943–945. [Google Scholar]
- Pignataro, G.; Simon, R.P.; Xiong, Z.G. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain 2007, 130, 151–158. [Google Scholar]
- Smith, H.S.; Deer, T.R.; Staats, P.S.; Singh, V.; Sehgal, N.; Cordner, H. Intrathecal drug delivery. Pain Physician 2008, 11, S89–S104. [Google Scholar]
- Kress, H.G.; Simpson, K.H.; Marchettini, P.; Ver Donck, A.; Varrassi, G. Intrathecal therapy: what has changed with the introduction of ziconotide. Pain Pract. 2009, 9, 338–347. [Google Scholar]
- Qadri, Y.J.; Berdiev, B.K.; Song, Y.; Lippton, H.L.; Fuller, C.M.; Benos, D.J. Psalmotoxin-1 docking to human acid-sensing ion channel-1. J. Biol. Chem. 2009, 284, 17625–17633. [Google Scholar]
- Pietra, F. Docking and MD simulations of the interaction of the tarantula peptide psalmotoxin-1 with ASIC1a channels using a homology model. J. Chem. Inf. Model. 2009, 49, 972–977. [Google Scholar]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4, 207. [Google Scholar]
- Krafte, D.S.; Bannon, A.W. Sodium channels and nociception: Recent concepts and therapeutic opportunities. Current Opin. Pharmacol. 2008, 8, 50–56. [Google Scholar]
- Priest, B.T. Future potential and status of selective sodium channel blockers for the treatment of pain. Curr. Opin. Drug Discov. Devel. 2009, 12, 682–692. [Google Scholar]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; Wu, G.; Jin, J.; Ding, B.; Zhu, X.; Shen, Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; Rees, M. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; Al-Gazali, L.; Hamamy, H.; Valente, E.M.; Gorman, S.; Williams, R.; McHale, D.P.; Wood, J.N.; Gribble, F.M.; Woods, C.G. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [PubMed]
- Goldberg, Y.P.; MacFarlane, J.; MacDonald, M.L.; Thompson, J.; Dube, M.P.; Mattice, M.; Fraser, R.; Young, C.; Hossain, S.; Pape, T.; Payne, B.; Radomski, C.; Donaldson, G.; Ives, E.; Cox, J.; Younghusband, H.B.; Green, R.; Duff, A.; Boltshauser, E.; Grinspan, G.A.; Dimon, J.H.; Sibley, B.G.; Andria, G.; Toscano, E.; Kerdraon, J.; Bowsher, D.; Pimstone, S.N.; Samuels, M.E.; Sherrington, R.; Hayden, M.R. Loss-of-function mutations in the NaV1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 2007, 71, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, K.B.; Nicholas, A.K.; Woods, C.G.; Mellgren, S.I.; Nebuchennykh, M.; Aasly, J. Two novel SCN9A mutations causing insensitivity to pain. Pain 2009, 143, 155–158. [Google Scholar]
- Clare, J.J. Targeting voltage-gated sodium channels for pain therapy. Expert Opin. Invest. Drugs 2010, 19, 45–62. [Google Scholar]
- Maggio, F.; Sollod, B.L.; Tedford, H.W.; Herzig, V.; King, G.F. Spider toxins and their potential for insect control. In Insect Pharmacology: Channels, Receptors, Toxins and Enzymes; Gilbert, L.I., Gill, S.S., Eds.; Academic Press: London, UK, 2010; pp. 101–123. [Google Scholar]
- Middleton, R.E.; Warren, V.A.; Kraus, R.L.; Hwang, J.C.; Liu, C.J.; Dai, G.; Brochu, R.M.; Kohler, M.G.; Gao, Y.D.; Garsky, V.M.; Bogusky, M.J.; Mehl, J.T.; Cohen, C.J.; Smith, M.M. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 2002, 41, 14734–14747. [Google Scholar]
- Schmalhofer, W.A.; Calhoun, J.; Burrows, R.; Bailey, T.; Kohler, M.G.; Weinglass, A.B.; Kaczorowski, G.J.; Garcia, M.L.; Koltzenburg, M.; Priest, B.T. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 2008, 74, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Lampe, R.A. Analgesic peptides from the venom of Grammostola spatulata. U.S. Patent 5,877,026, 2 March 1999. [Google Scholar]
- Khakh, B.S.; North, R.A. P2X receptors as cell-surface ATP sensors in health and disease. Nature 2006, 442, 527–532. [Google Scholar]
- Skaper, S.D.; Debetto, P.; Giusti, P. The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J. 2010, 24, 337–345. [Google Scholar]
- Nicke, A.; Baumert, H.G.; Rettinger, J.; Eichele, A.; Lambrecht, G.; Mutschler, E.; Schmalzing, G. P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. EMBO J. 1998, 17, 3016–3028. [Google Scholar]
- Wirkner, K.; Sperlagh, B.; Illes, P. P2X3 receptor involvement in pain states. Mol. Neurobiol. 2007, 36, 165–183. [Google Scholar]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar]
- Gunosewoyo, H.; Kassiou, M. P2X purinergic receptor ligands: recently patented compounds. Expert Opin. Ther. Pat. 2010, 20, 625–646. [Google Scholar]
- North, R.A. P2X3 receptors and peripheral pain mechanisms. J. Physiol. 2004, 554, 301–308. [Google Scholar]
- Jarvis, M.F.; Burgard, E.C.; McGaraughty, S.; Honore, P.; Lynch, K.; Brennan, T.J.; Subieta, A.; Van Biesen, T.; Cartmell, J.; Bianchi, B.; Niforatos, W.; Kage, K.; Yu, H.; Mikusa, J.; Wismer, C.T.; Zhu, C.Z.; Chu, K.; Lee, C.H.; Stewart, A.O.; Polakowski, J.; Cox, B.F.; Kowaluk, E.; Williams, M.; Sullivan, J.; Faltynek, C. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc. Natl. Acad. Sci. USA 2002, 99, 17179–17184. [Google Scholar]
- Hausmann, R.; Rettinger, J.; Gerevich, Z.; Meis, S.; Kassack, M.U.; Illes, P.; Lambrecht, G.; Schmalzing, G. The suramin analog 4,4',4'',4'''-(carbonylbis(imino-5,1,3-benzenetriylbis (carbonylimino)))tetra-kis-benzenesulfonic acid (NF110) potently blocks P2X3 receptors: Subtype selectivity is determined by location of sulfonic acid groups. Mol. Pharmacol. 2006, 69, 2058–2067. [Google Scholar]
- Jung, K.Y.; Moon, H.D.; Lee, G.E.; Lim, H.H.; Park, C.S.; Kim, Y.C. Structure-activity relationship studies of spinorphin as a potent and selective human P2X(3) receptor antagonist. J. Med. Chem. 2007, 50, 4543–4547. [Google Scholar]
- Miljanich, G.P. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar]
- Sharpe, I.A.; Gehrmann, J.; Loughnan, M.L.; Thomas, L.; Adams, D.A.; Atkins, A.; Palant, E.; Craik, D.J.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Two new classes of conopeptides inhibit the α1-adrenoceptor and noradrenaline transporter. Nat. Neurosci. 2001, 4, 902–907. [Google Scholar]
- Patapoutian, A.; Tate, S.; Woolf, C.J. Transient receptor potential channels: Targeting pain at the source. Nat. Rev. Drug Discov. 2009, 8, 55–68. [Google Scholar]
- Cortright, D.N.; Szallasi, A. TRP channels and pain. Curr. Pharm. Des. 2009, 15, 1736–1749. [Google Scholar]
- Martinac, B.; Kloda, A. Evolutionary origins of mechanosensitive ion channels. Prog. Biophys. Mol. Biol. 2003, 82, 11–24. [Google Scholar]
- Sachs, F. Stretch-activated ion channels: What are they? Physiology 2010, 25, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Suchyna, T.M.; Johnson, J.H.; Hamer, K.; Leykam, J.F.; Gage, D.A.; Clemo, H.F.; Baumgarten, C.M.; Sachs, F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 2000, 115, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Oswald, R.E.; Suchyna, T.M.; McFeeters, R.; Gottlieb, P.; Sachs, F. Solution structure of peptide toxins that block mechanosensitive ion channels. J. Biol. Chem. 2002, 277, 34443–34450. [Google Scholar]
- Bode, F.; Sachs, F.; Franz, M.R. Tarantula peptide inhibits atrial fibrillation. Nature 2001, 409, 35–36. [Google Scholar]
- Suchyna, T.M.; Tape, S.E.; Koeppe, R.E., 2nd; Andersen, O.S.; Sachs, F.; Gottlieb, P.A. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 2004, 430, 235–240. [Google Scholar] [PubMed]
- Garcia, M.L. Ion channels: Gate expectations. Nature 2004, 430, 153–155. [Google Scholar]
- Jung, H.J.; Kim, P.I.; Lee, S.K.; Lee, C.W.; Eu, Y.J.; Lee, D.G.; Earm, Y.E.; Kim, J.I. Lipid membrane interaction and antimicrobial activity of GsMTx-4, an inhibitor of mechanosensitive channel. Biochem. Biophys. Res. Commun. 2006, 340, 633–638. [Google Scholar]
- Andersson, K.E.; Wagner, G. Physiology of penile erection. Physiol. Rev. 1995, 75, 191–236. [Google Scholar]
- Toda, N.; Ayajiki, K.; Okamura, T. Nitric oxide and penile erectile function. Pharmacol. Therapeut. 2005, 106, 233–266. [Google Scholar]
- Fusco, F.; Razzoli, E.; Imbimbo, C.; Rossi, A.; Verze, P.; Mirone, V. A new era in the treatment of erectile dysfunction: Chronic phosphodiesterase type 5 inhibition. BJU Int. 2010, 105, 1634–1639. [Google Scholar]
- Webb, D.J.; Freestone, S.; Allen, M.J.; Muirhead, G.J. Sildenafil citrate and blood-pressure-lowering drugs: Results of drug interaction studies with an organic nitrate and a calcium antagonist. Am. J. Cardiol. 1999, 83, 21–28. [Google Scholar]
- Wright, P.J. Comparison of phosphodiesterase type 5 (PDE5) inhibitors. Int. J. Clin. Pract. 2006, 60, 967–975. [Google Scholar]
- Cordeiro, M.N.; Diniz, C.R.; Valentim, A.C.; von Eickstedt, V.R.D.; Gilroy, J.; Richardson, M. The purification and amino acid sequences of four Tx2 neurotoxins from the venom of the Brazilian 'armed' spider Phoneutria nigriventer (Keys). FEBS Lett. 1992, 310, 153–156. [Google Scholar]
- Matavel, A.; Fleury, C.; Oliveira, L.C.; Molina, F.; de Lima, M.E.; Cruz, J.S.; Cordeiro, M.N.; Richardson, M.; Ramos, C.H.; Beirao, P.S. Structure and activity analysis of two spider toxins that alter sodium channel inactivation kinetics. Biochemistry 2009, 48, 3078–3088. [Google Scholar]
- Nunes, K.P.; Costa-Goncalves, A.; Lanza, L.F.; Cortes, S.F.; Cordeiro, M.N.; Richardson, M.; Pimenta, A.M.; Webb, R.C.; Leite, R.; De Lima, M.E. Tx2-6 toxin of the Phoneutria nigriventer spider potentiates rat erectile function. Toxicon 2008, 51, 1197–1206. [Google Scholar]
- Andrade, E.; Villanova, F.; Borra, P.; Leite, K.; Troncone, L.; Cortez, I.; Messina, L.; Paranhos, M.; Claro, J.; Srougi, M. Penile erection induced in vivo by a purified toxin from the Brazilian spider Phoneutria nigriventer. BJU Int. 2008, 102, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Platnick, N.I. Advances in Spider Taxonomy,1992–1995: With Redescriptions 1940–1980; New York Entomological Society & The American Museum of Natural History: New York, NY, USA, 1997. [Google Scholar]
- Bi, P.; Whitby, M.; Walker, S.; Parton, K.A. Trends in mortality rates for infectious and parasitic diseases in Australia: 1907–1997. Intern. Med. J. 2003, 33, 152–162. [Google Scholar]
- Alanis, A.J. Resistance to antibiotics: Are we in the post-antibiotic era? Arch Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Robbel, L.; Marahiel, M.A. Daptomycin, a bacterial lipopeptide synthesized by a nonribosomal machinery. J. Biol. Chem. 2010, 285, 27501–27508. [Google Scholar]
- Vooturi, S.K.; Firestine, S.M. Synthetic membrane-targeted antibiotics. Curr. Med. Chem. 2010, 17, 2292–2300. [Google Scholar]
- Yan, L.; Adams, M.E. Lycotoxins, antimicrobial peptides from venom of the wolf spider Lycosa carolinensis. J. Biol. Chem. 1998, 273, 2059–2066. [Google Scholar]
- Kuhn-Nentwig, L.; Muller, J.; Schaller, J.; Walz, A.; Dathe, M.; Nentwig, W. Cupiennin 1, a new family of highly basic antimicrobial peptides in the venom of the spider Cupiennius salei (Ctenidae). J. Biol. Chem. 2002, 277, 11208–11216. [Google Scholar]
- Ballweber, P.; Markl, J.; Burmester, T. Complete hemocyanin subunit sequences of the hunting spider Cupiennius salei: Recent hemocyanin remodeling in entelegyne spiders. J. Biol. Chem. 2002, 277, 14451–14457. [Google Scholar] [PubMed][Green Version]
- Kuhn-Nentwig, L. Antimicrobial and cytolytic peptides of venomous arthropods. Cell. Mol. Life Sci. 2003, 60, 2651–2668. [Google Scholar]
- Nomura, K.; Corzo, G. The effect of binding of spider-derived antimicrobial peptides, oxyopinins, on lipid membranes. Biochim. Biophys. Acta 2006, 1758, 1475–1482. [Google Scholar]
- Pukala, T.L.; Boland, M.P.; Gehman, J.D.; Kuhn-Nentwig, L.; Separovic, F.; Bowie, J.H. Solution structure and interaction of cupiennin 1a, a spider venom peptide, with phospholipid bilayers. Biochemistry 2007, 46, 3576–3585. [Google Scholar] [PubMed]
- Dubovskii, P.; Volynsky, P.E.; Polyansky, A.A.; Karpunin, D.V.; Chupin, V.V.; Efremov, R.G.; Arseniev, A.S. Three-dimensional structure/hydrophobicity of latarcins specifies their mode of membrane activity. Biochemistry 2008, 47, 3525–3233. [Google Scholar]
- Vorontsova, O.V.; Egorova, N.S.; Arseniev, A.S.; Feofanov, A.V. Haemolytic and cytotoxic action of latarcin Ltc2a. Biochimie 2010. [Google Scholar]
- Corzo, G.; Villegas, E.; Gomez-Lagunas, F.; Possani, L.D.; Belokoneva, O.S.; Nakajima, T. Oxyopinins, large amphipathic peptides isolated from the venom of the wolf spider Oxyopes kitabensis with cytolytic properties and positive insecticidal cooperativity with spider neurotoxins. J. Biol. Chem. 2002, 277, 23627–23637. [Google Scholar] [PubMed]
- Kuhn-Nentwig, L.; Schaller, J.; Nentwig, W. Biochemistry, toxicology and ecology of the venom of the spider Cupiennius salei (Ctenidae). Toxicon 2004, 43, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Adão, R.; Seixas, R.; Gomes, P.; Pessoa, J.C.; Bastos, M. Membrane structure and interactions of a short Lycotoxin I analogue. J. Pept. Sci. 2008, 14, 528–534. [Google Scholar]
- Henriques, S.T.; Craik, D.J. Cyclotides as templates in drug design. Drug Discov. Today 2010, 15, 57–64. [Google Scholar]
- World Health Organization, World Malaria Report 2009; WHO Press: Geneva, Switzerland, 2009.
- Enayati, A.; Hemingway, J. Malaria management: past, present, and future. Annu. Rev. Entomol. 2010, 55, 569–591. [Google Scholar] [CrossRef] [PubMed]
- Staines, H.M.; Ellory, J.C.; Chibale, K. The new permeability pathways: targets and selective routes for the development of new antimalarial agents. Comb. Chem. High Throughput Screen 2005, 8, 81–88. [Google Scholar]
- Nicholson, G.M.; Graudins, A. Spiders of medical importance in the Asia-Pacific: atracotoxin, latrotoxin and related spider neurotoxins. Clin. Exp. Pharmacol. Physiol. 2002, 29, 785–794. [Google Scholar]
- Craik, D.J.; Adams, D.J. Chemical modification of conotoxins to improve stability and activity. ACS Chem. Biol. 2007, 2, 457–468. [Google Scholar]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar]
- Knauf, M.J.; Bell, D.P.; Hirtzer, P.; Luo, Z.P.; Young, J.D.; Katre, N.V. Relationship of effective molecular-size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. J. Biol. Chem. 1988, 263, 15064–15070. [Google Scholar]
- Pettit, D.K.; Gombotz, W.R. The development of site-specific drug-delivery systems for protein and peptide biopharmaceuticals. Trends Biotechnol. 1998, 16, 343–349. [Google Scholar]
- Tedford, H.W.; Fletcher, J.I.; King, G.F. Functional significance of the β-hairpin in the insecticidal neurotoxin ω-atracotoxin-Hv1a. J. Biol. Chem. 2001, 276, 26568–26576. [Google Scholar]
- Tedford, H.W.; Gilles, N.; Ménez, A.; Doering, C.J.; Zamponi, G.W.; King, G.F. Scanning mutagenesis of ω-atracotoxin-Hv1a reveals a spatially restricted epitope that confers selective activity against invertebrate calcium channels. J. Biol. Chem. 2004, 279, 44133–44140. [Google Scholar]
- Gadek, T.R.; Nicholas, J.B. Small molecule antagonists of proteins. Biochem. Pharmacol. 2003, 65, 1–8. [Google Scholar]
- Baell, J.B.; Duggan, P.J.; Lok, Y.P. ω-Conotoxins and approaches to their nonpeptide mimetics. Aust. J. Chem. 2004, 57, 179–185. [Google Scholar]
- Harvey, A.J.; Gable, R.W.; Baell, J.B. A three-residue, continuous binding epitope peptidomimetic of ShK toxin as a Kv1.3 inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 3193–3196. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-Venom Peptides as Therapeutics. Toxins 2010, 2, 2851-2871. https://doi.org/10.3390/toxins2122851
Saez NJ, Senff S, Jensen JE, Er SY, Herzig V, Rash LD, King GF. Spider-Venom Peptides as Therapeutics. Toxins. 2010; 2(12):2851-2871. https://doi.org/10.3390/toxins2122851
Chicago/Turabian StyleSaez, Natalie J., Sebastian Senff, Jonas E. Jensen, Sing Yan Er, Volker Herzig, Lachlan D. Rash, and Glenn F. King. 2010. "Spider-Venom Peptides as Therapeutics" Toxins 2, no. 12: 2851-2871. https://doi.org/10.3390/toxins2122851
APA StyleSaez, N. J., Senff, S., Jensen, J. E., Er, S. Y., Herzig, V., Rash, L. D., & King, G. F. (2010). Spider-Venom Peptides as Therapeutics. Toxins, 2(12), 2851-2871. https://doi.org/10.3390/toxins2122851