Lipopolysaccharide Induces Mitochondrial Fragmentation and Energetic Shift in Reactive Microglia: Evidence for a Cell-Autonomous Program of Metabolic Plasticity

 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
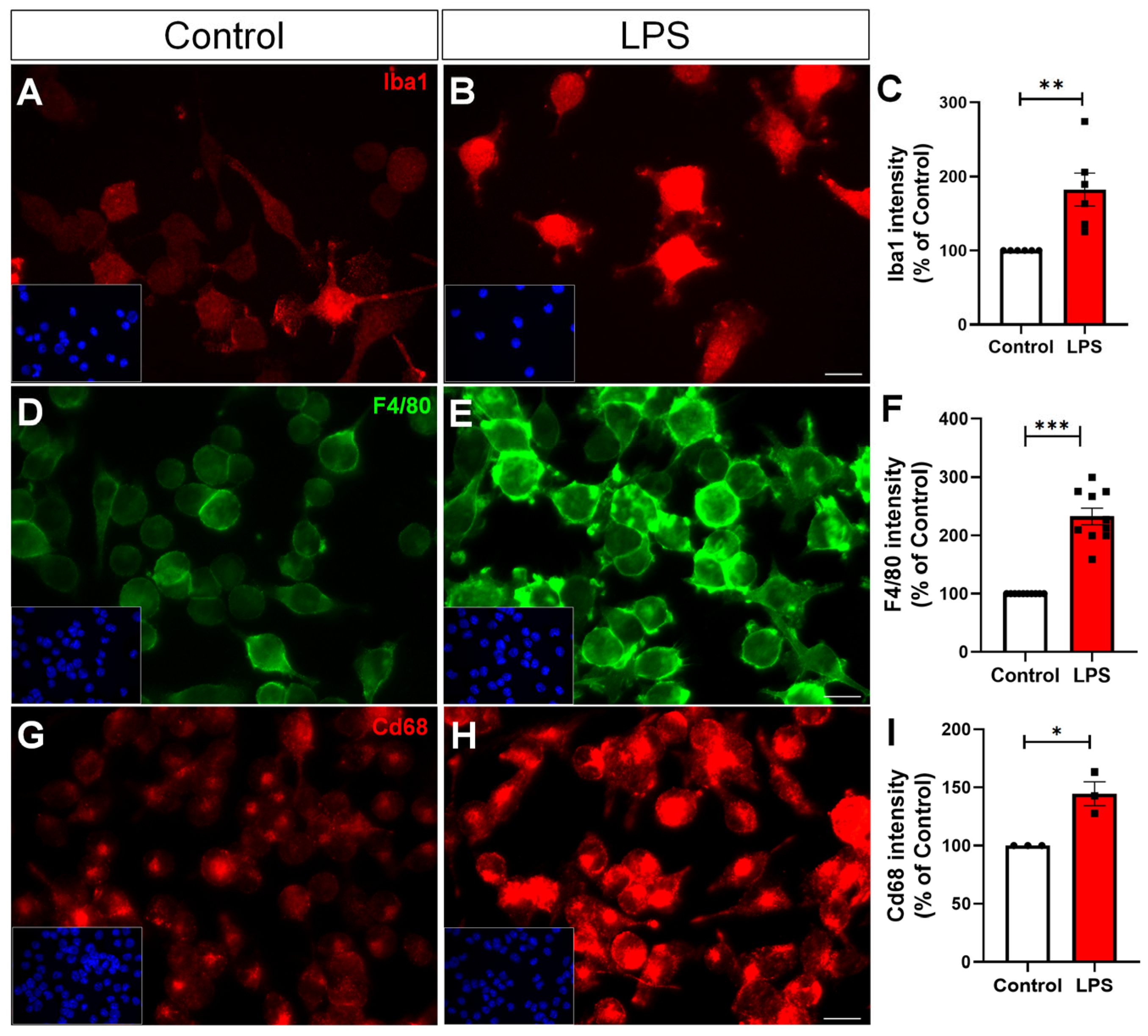
2.1. LPS Induces Microglial Reactivity
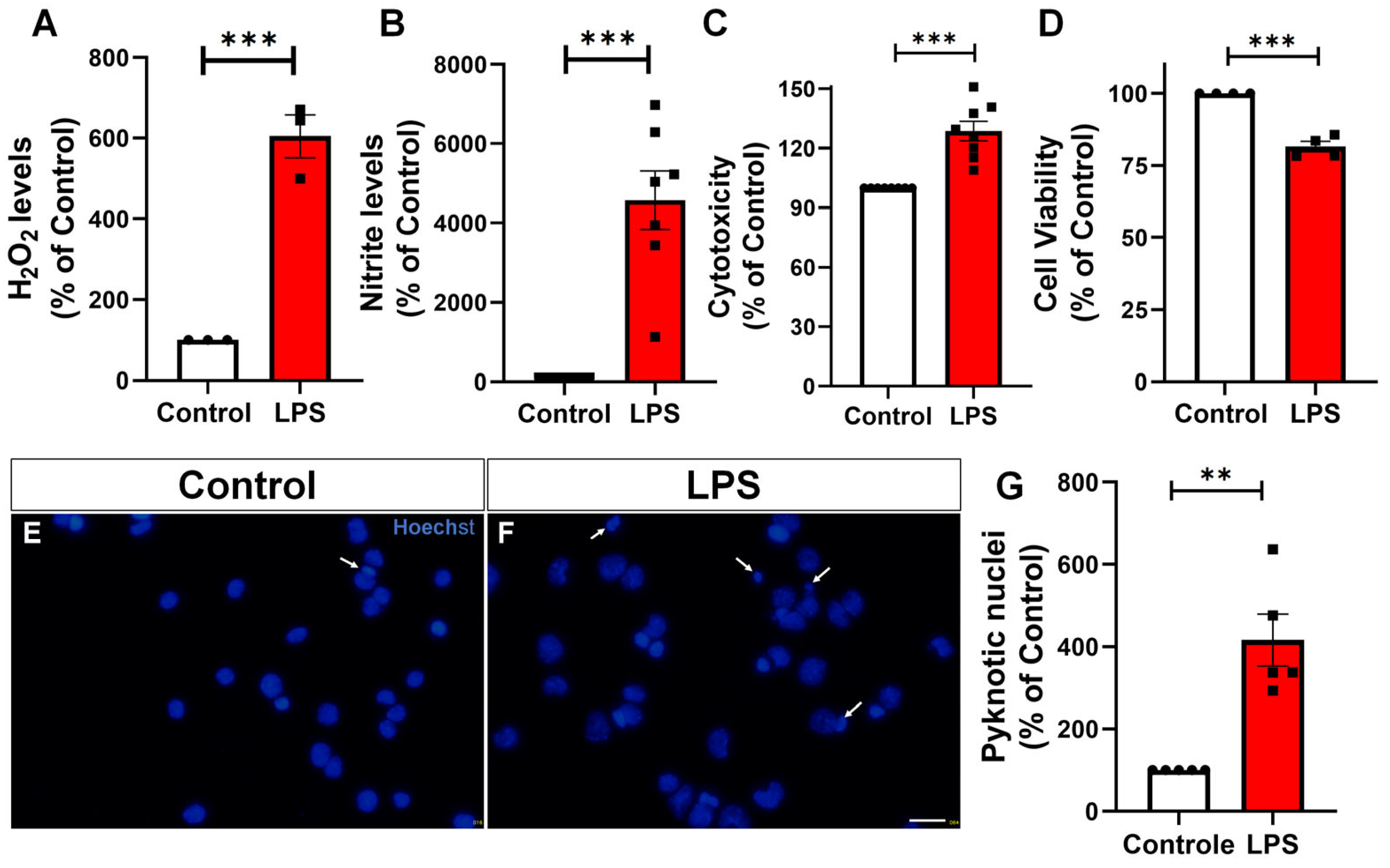
2.2. LPS Increases H2O2, Nitric Oxide Production, Cytotoxicity, and Apoptosis in Microglia
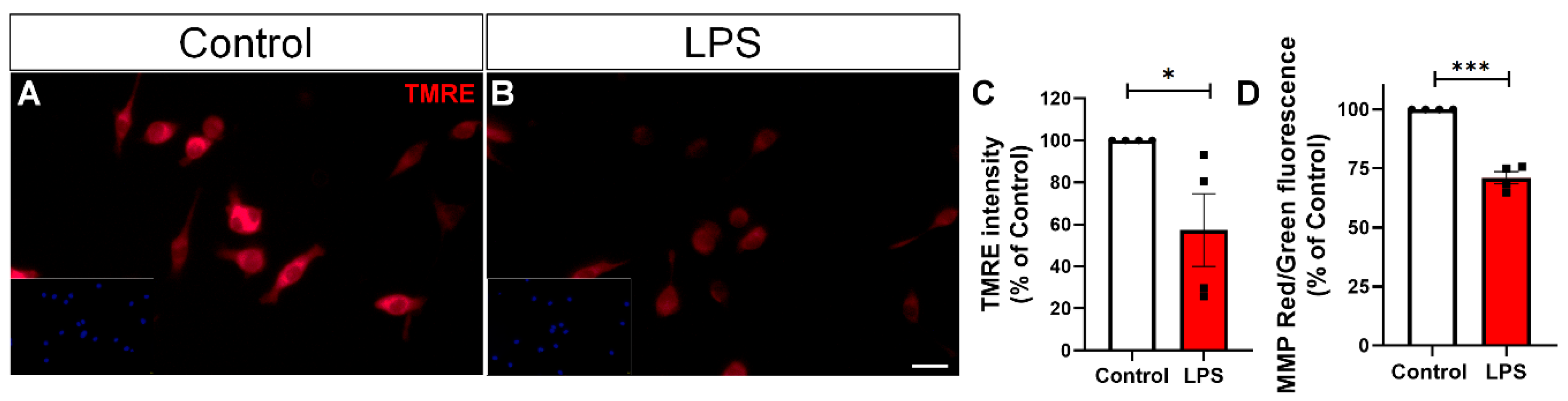
2.3. LPS Triggers Mitochondrial Membrane Potential Loss and Mitochondrial Fission in BV-2 Microglial Cells
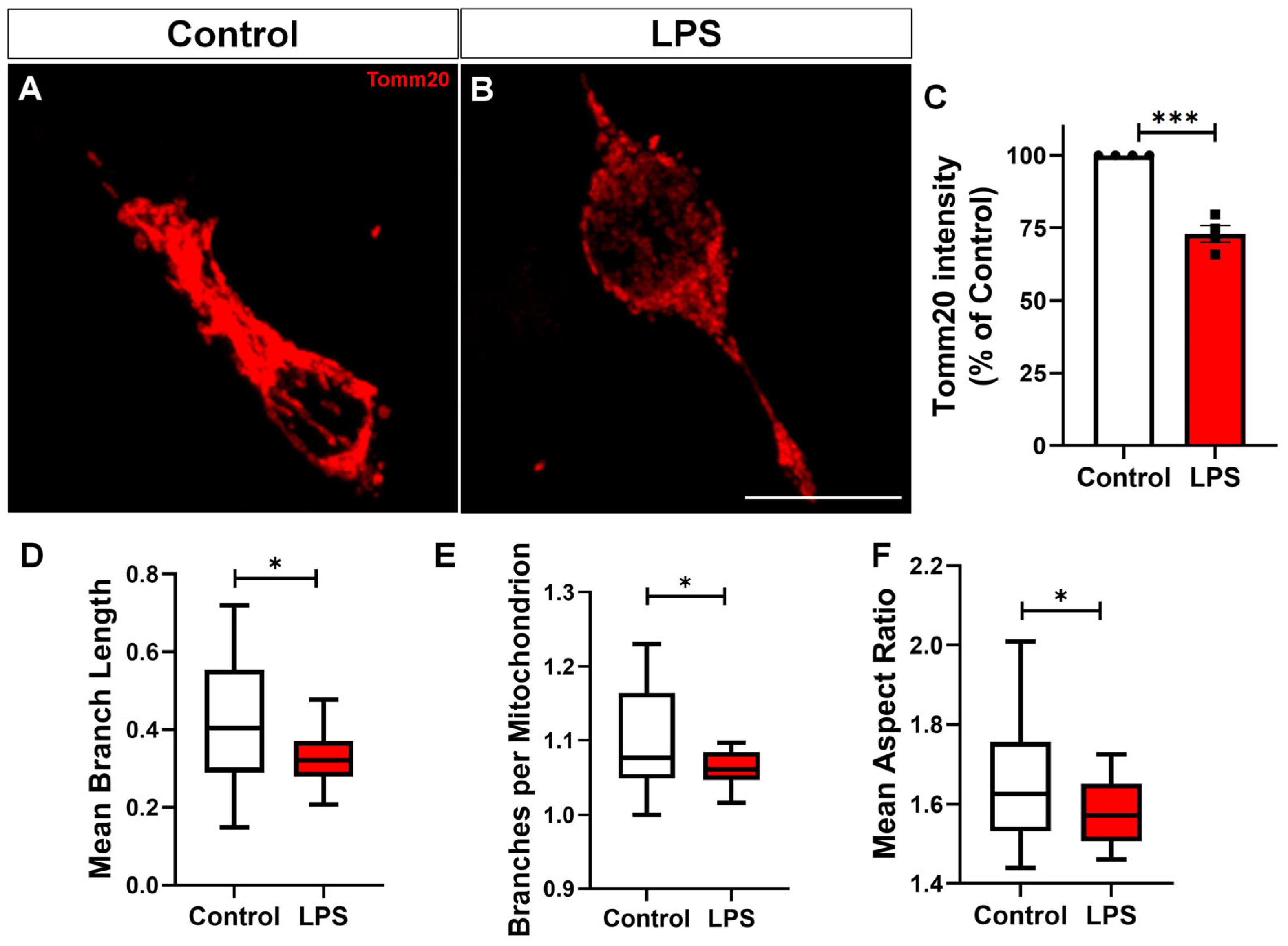
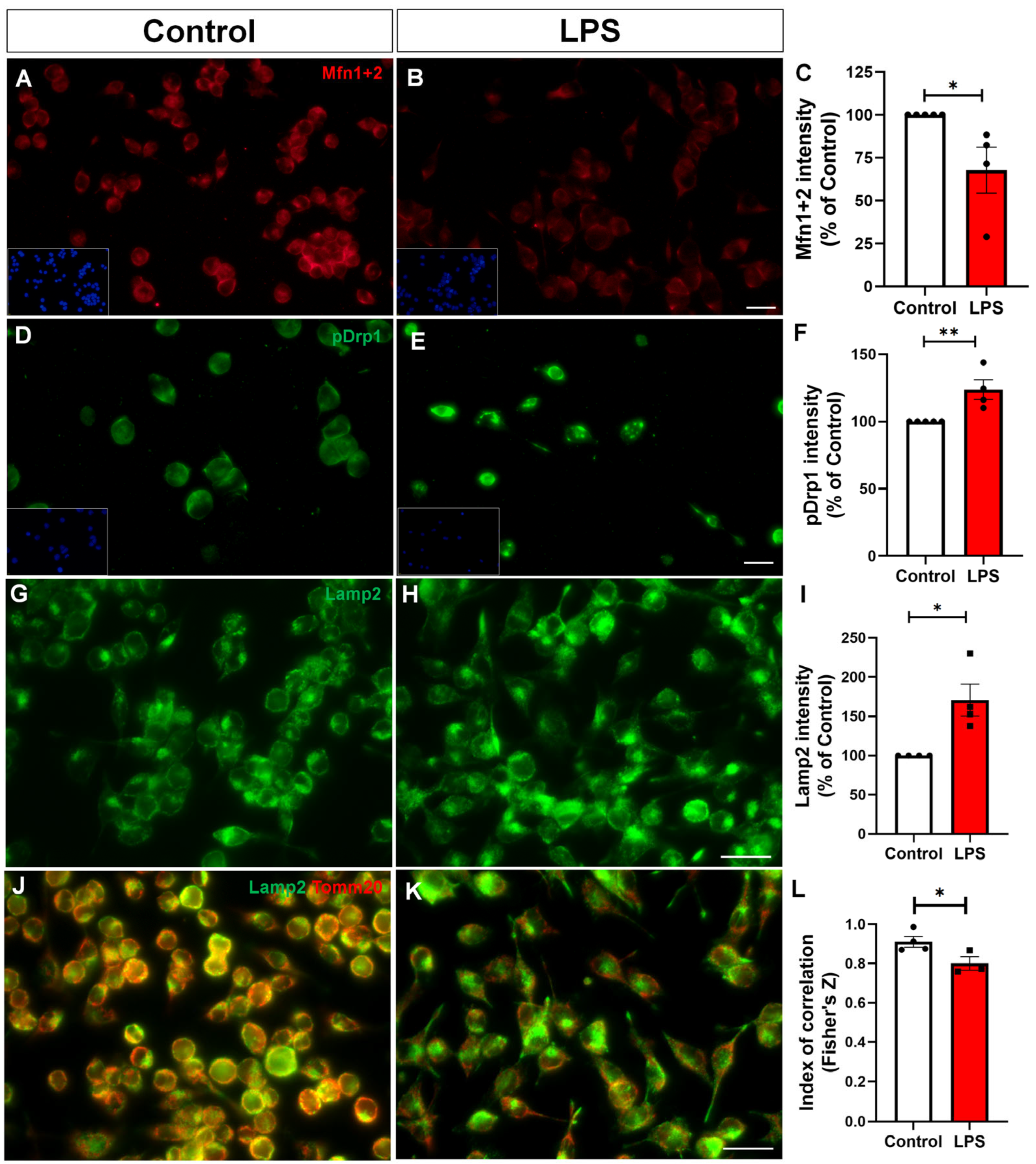
2.4. Reactive Microglia Exhibit Altered Mitochondrial Dynamics and Impaired Mitophagy
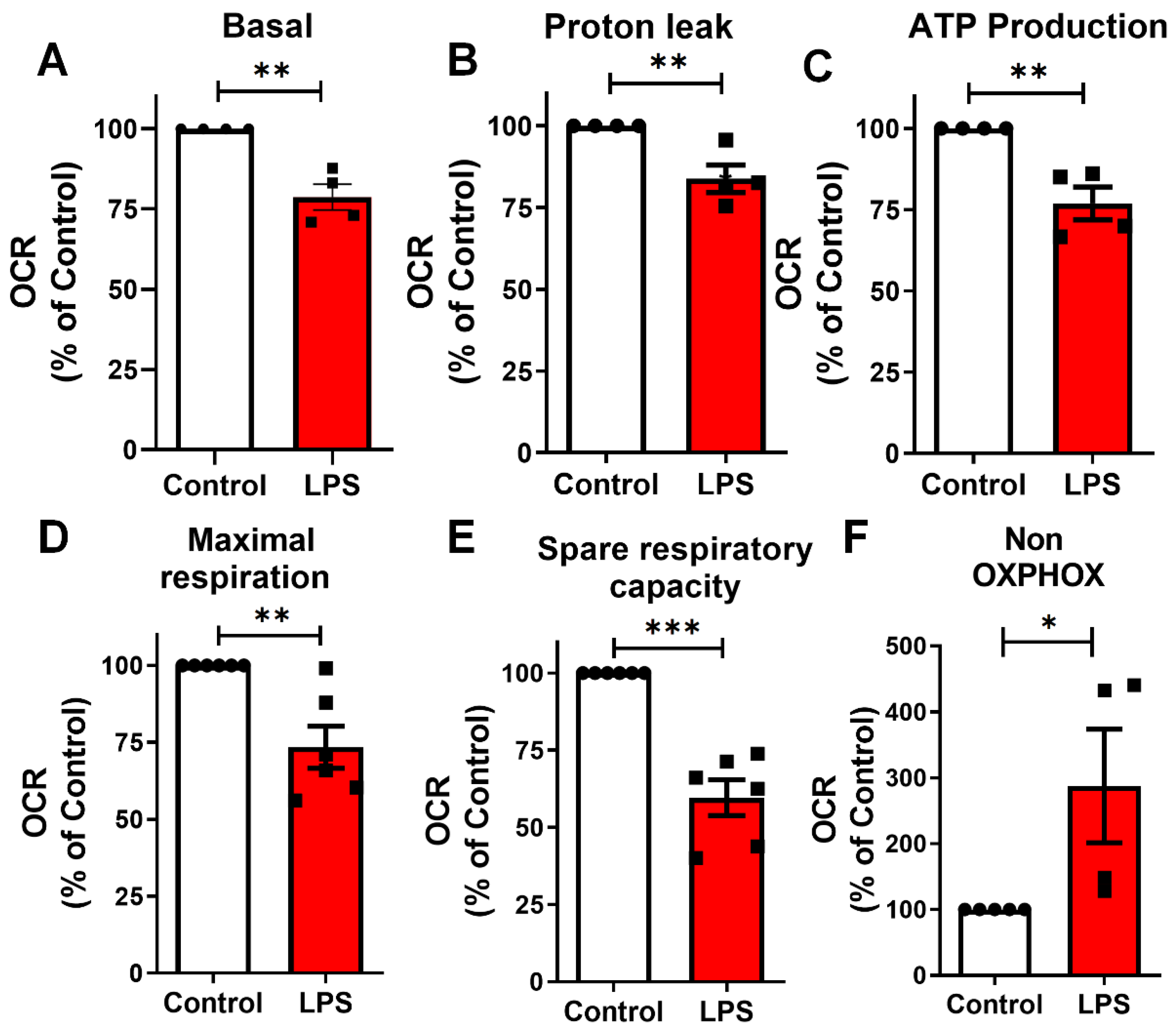
2.5. LPS Induces Mitochondrial Bioenergetic Shift in BV-2 Microglia
2.6. LPS-Induced Alterations in Glycolytic Flux and Impaired ATP Maintenance in Microglia
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Treatment
5.3. Immunocytochemistry
5.4. Immunofluorescence Analysis
5.5. Measurement of Extracellular Hydrogen Peroxide
5.6. Cell Viability Assessment
5.7. Extracellular Lactate Levels
5.8. Nitrite Levels
5.9. Intracellular ATP Levels
5.10. Assessment of Mitochondrial Membrane Potential Using TMRE and JC-1 Dyes
5.11. Mitochondrial Respiration in Microglial Cultures
5.12. MTT Assay for Cell Viability
5.13. Data and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cd68 | Cluster of differentiation 68 |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| DAMPs | Danger-associated molecular patterns |
| DMEM | Dulbecco’s Modified Eagle Medium |
| H2O2 | Hydrogen peroxide |
| IBA1 | Ionized calcium-binding adaptor molecule 1 |
| JC-1 | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide |
| Lamp2 | Lysosome-associated membrane protein 2 |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| Mfn1 | Mitofusin 1 |
| Mfn2 | Mitofusin 2 |
| MiNA | Mitochondrial Network Analysis |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NO | Nitric oxide |
| NOX2 | NADPH oxidase 2 |
| OXPHOS | Oxidative phosphorylation |
| p-DRP | Phosphorylated dynamin-related protein 1 at serine 616 |
| ROS | Reactive oxygen species |
| TLR4 | Toll-like receptor 4 |
| TMRE | Tetramethylrhodamine ethyl ester |
| Tomm20 | Translocase of outer mitochondrial membrane 20 |
References
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [PubMed]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Dai, X.J.; Li, N.; Yu, L.; Chen, Z.Y.; Hua, R.; Qin, X.; Zhang, Y.M. Activation of BV2 microglia by lipopolysaccharide triggers an inflammatory reaction in PC12 cell apoptosis through a toll-like receptor 4-dependent pathway. Cell Stress Chaperones 2015, 20, 321–331. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Ana, B. Aged-Related Changes in Microglia and Neurodegenerative Diseases: Exploring the Connection. Biomedicines 2024, 12, 1737. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef]
- Montilla, A.; Ruiz, A.; Marquez, M.; Sierra, A.; Matute, C.; Domercq, M. Role of Mitochondrial Dynamics in Microglial Activation and Metabolic Switch. ImmunoHorizons 2021, 5, 615–626. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Willis, C.M.; Krzak, G.; Hamel, R.; Pirvan, L.; Ionescu, R.B.; Reisz, J.A.; Prag, H.A.; Garcia-Segura, M.E.; Wu, V.; et al. Mitochondrial complex I activity in microglia sustains neuroinflammation. Nature 2024, 628, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Voloboueva, L.A.; Emery, J.F.; Sun, X.; Giffard, R.G. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013, 587, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, R.; Xu, Z.; Ke, Y.; Sun, R.; Yang, H.; Zhang, X.; Zhen, X.; Zheng, L.T. Early glycolytic reprogramming controls microglial inflammatory activation. J. Neuroinflamm. 2021, 18, 129. [Google Scholar] [CrossRef] [PubMed]
- Pereira, O.R., Jr.; Ramos, V.M.; Cabral-Costa, J.V.; Kowaltowski, A.J. Changes in mitochondrial morphology modulate LPS-induced loss of calcium homeostasis in BV-2 microglial cells. J. Bioenerg. Biomembr. 2021, 53, 109–118. [Google Scholar] [CrossRef]
- Hunter, R.; Ojha, U.; Bhurtel, S.; Bing, G.; Choi, D.Y. Lipopolysaccharide-induced functional and structural injury of the mitochondria in the nigrostriatal pathway. Neurosci. Res. 2017, 114, 62–69. [Google Scholar] [CrossRef]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond Activation: Characterizing Microglial Functional Phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef]
- Gao, F.; Zheng, Z.; Liu, X.; Li, J. CMPK2 promotes microglial activation through the cGAS-STING pathway in the neuroinflammatory mechanism. Sci. Rep. 2025, 15, 11807. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Hou, X.O.; Hu, L.F. Roles of microglial mitophagy in neurological disorders. Front. Aging Neurosci. 2022, 14, 979869. [Google Scholar] [CrossRef]
- Qin, P.; Sun, Y.; Li, L. Mitochondrial dysfunction in chronic neuroinflammatory diseases (Review). Int. J. Mol. Med. 2024, 53, 47. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- She, Y.; Shao, C.Y.; Liu, Y.F.; Huang, Y.; Yang, J.; Wan, H.T. Catalpol reduced LPS induced BV2 immunoreactivity through NF-kappaB/NLRP3 pathways: An in Vitro and in silico study. Front. Pharmacol. 2024, 15, 1415445. [Google Scholar] [CrossRef]
- Chang, R.C.; Chen, W.; Hudson, P.; Wilson, B.; Han, D.S.; Hong, J.S. Neurons reduce glial responses to lipopolysaccharide (LPS) and prevent injury of microglial cells from over-activation by LPS. J. Neurochem. 2001, 76, 1042–1049. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef]
- Monsorno, K.; Buckinx, A.; Paolicelli, R.C. Microglial metabolic flexibility: Emerging roles for lactate. Trends Endocrinol. Metab. TEM 2022, 33, 186–195. [Google Scholar] [CrossRef]
- Sangineto, M.; Ciarnelli, M.; Cassano, T.; Radesco, A.; Moola, A.; Bukke, V.N.; Romano, A.; Villani, R.; Kanwal, H.; Capitanio, N.; et al. Metabolic reprogramming in inflammatory microglia indicates a potential way of targeting inflammation in Alzheimer’s disease. Redox Biol. 2023, 66, 102846. [Google Scholar] [CrossRef]
- Matias, I.; Diniz, L.P.; Damico, I.V.; Araujo, A.P.B.; Neves, L.D.S.; Vargas, G.; Leite, R.E.P.; Suemoto, C.K.; Nitrini, R.; Jacob-Filho, W.; et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 2022, 21, e13521. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.P.B.; Vargas, G.; Hayashide, L.S.; Matias, I.; Andrade, C.B.V.; de Carvalho, J.J.; Gomes, F.C.A.; Diniz, L.P. Aging promotes an increase in mitochondrial fragmentation in astrocytes. Front. Cell. Neurosci. 2024, 18, 1496163. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson’s disease model. Neuropharmacology 2022, 207, 108963. [Google Scholar] [CrossRef]
- Zhang, J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biol. 2015, 4, 242–259. [Google Scholar] [CrossRef]
- Du, D.; Hu, L.; Wu, J.; Wu, Q.; Cheng, W.; Guo, Y.; Guan, R.; Wang, Y.; Chen, X.; Yan, X.; et al. Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J. Neuroinflamm. 2017, 14, 169. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Bamburg, J.R.; Minamide, L.S.; Wiggan, O.; Tahtamouni, L.H.; Kuhn, T.B. Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration. Cells 2021, 10, 2726. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef]
- Sancho, P.; Martin-Sanz, P.; Fabregat, I. Reciprocal regulation of NADPH oxidases and the cyclooxygenase-2 pathway. Free Radic. Biol. Med. 2011, 51, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wang, D.; Qu, Y.; Li, J.; An, K.; Mao, Z.; Li, J.; Xiong, Y.; Min, Z.; Xue, Z. Enhanced glycolysis-derived lactate promotes microglial activation in Parkinson’s disease via histone lactylation. npj Park. Dis. 2025, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yu, W.; Duan, W.; Wang, T.; Liu, Y. Dexmedetomidine attenuates LPS-mediated BV2 microglia cells inflammation via inhibition of glycolysis. Fundam. Clin. Pharmacol. 2020, 34, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Liu, P.; Deng, Y.; Li, J.; Zhao, S. L- lactate inhibits lipopolysaccharide-induced inflammation of microglia in the hippocampus. Int. J. Neurosci. 2024, 134, 45–52. [Google Scholar] [CrossRef]
- Li, Y.; Xia, X.; Wang, Y.; Zheng, J.C. Mitochondrial dysfunction in microglia: A novel perspective for pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2022, 19, 248. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Wang, J.; Guo, X.; Song, Y.; Fu, K.; Gao, Z.; Liu, D.; He, W.; Yang, L.L. Energy metabolism in health and diseases. Signal Transduct. Target. Ther. 2025, 10, 69. [Google Scholar] [CrossRef]
- He, Y.; Yao, X.; Taylor, N.; Bai, Y.; Lovenberg, T.; Bhattacharya, A. RNA sequencing analysis reveals quiescent microglia isolation methods from postnatal mouse brains and limitations of BV2 cells. J. Neuroinflamm. 2018, 15, 153. [Google Scholar] [CrossRef]
- Tawbeh, A.; Raas, Q.; Tahri-Joutey, M.; Keime, C.; Kaiser, R.; Trompier, D.; Nasser, B.; Bellanger, E.; Dessard, M.; Hamon, Y.; et al. Immune response of BV-2 microglial cells is impacted by peroxisomal beta-oxidation. Front. Mol. Neurosci. 2023, 16, 1299314. [Google Scholar] [CrossRef]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef]
- Jin, M.; Kim, B.W.; Koppula, S.; Kim, I.S.; Park, J.H.; Kumar, H.; Choi, D.K. Molecular effects of activated BV-2 microglia by mitochondrial toxin 1-methyl-4-phenylpyridinium. Neurotoxicology 2012, 33, 147–155. [Google Scholar] [CrossRef]
- Lin, M.M.; Liu, N.; Qin, Z.H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Gorlewicz, A.; Krawczyk, K.; Szczepankiewicz, A.A.; Trzaskoma, P.; Mulle, C.; Wilczynski, G.M. Colocalization Colormap—An ImageJ Plugin for the Quantification and Visualization of Colocalized Signals. Neuroinformatics 2020, 18, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Leser, F.S.; Junyor, F.S.; Pagnoncelli, I.B.; Delgado, A.B.; Medeiros, I.; Nobrega, A.C.C.; Andrade, B.D.S.; de Lima, M.N.; da Silva, N.E.; Jacob, L.; et al. CCL21-CCR7 blockade prevents neuroinflammation and degeneration in Parkinson’s disease models. J. Neuroinflamm. 2025, 22, 31. [Google Scholar] [CrossRef]
- Diniz, L.P.; Morgado, J.; Bergamo Araujo, A.P.; da Silva Antonio, L.M.; Mota-Araujo, H.P.; de Sena Murteira Pinheiro, P.; Sagrillo, F.S.; Cesar, G.V.; Ferreira, S.T.; Figueiredo, C.P.; et al. Histone deacetylase inhibition mitigates cognitive deficits and astrocyte dysfunction induced by amyloid-beta (Abeta) oligomers. Br. J. Pharmacol. 2024, 181, 4028–4049. [Google Scholar] [CrossRef]
- Diniz, L.P.; Matias, I.; Araujo, A.P.B.; Garcia, M.N.; Barros-Aragao, F.G.Q.; Alves-Leon, S.V.; de Souza, J.M.; Foguel, D.; Figueiredo, C.P.; Braga, C.; et al. alpha-synuclein oligomers enhance astrocyte-induced synapse formation through TGF-beta1 signaling in a Parkinson’s disease model. J. Neurochem. 2019, 150, 138–157. [Google Scholar] [CrossRef]
- Assis-de-Lemos, G.; Monteiro, J.; Oliveira-Valenca, V.M.; Melo, G.A.; Reis, R.A.M.; Rehen, S.K.; Silveira, M.S.; Galina, A. Dopamine signaling impairs ROS modulation by mitochondrial hexokinase in human neural progenitor cells. Biosci. Rep. 2021, 41, BSR20211191. [Google Scholar] [CrossRef]
- Assis-de-Lemos, G.; Ledur, P.F.; Karmirian, K.; Rehen, S.K.; Galina, A. A Protocol to Study Mitochondrial Function in Human Neural Progenitors and iPSC-Derived Astrocytes. Curr. Protoc. Toxicol. 2020, 85, e97. [Google Scholar] [CrossRef]
- Diniz, L.P.; Araujo, A.P.B.; Carvalho, C.F.; Matias, I.; de Sa Hayashide, L.; Marques, M.; Pessoa, B.; Andrade, C.B.V.; Vargas, G.; Queiroz, D.D.; et al. Accumulation of damaged mitochondria in aging astrocytes due to mitophagy dysfunction: Implications for susceptibility to mitochondrial stress. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167470. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dos Santos, M.P.; Leocadio, V.E.; de Sá Hayashide, L.; Marques, M.; Carvalho, C.F.; Galina, A.; Diniz, L.P. Lipopolysaccharide Induces Mitochondrial Fragmentation and Energetic Shift in Reactive Microglia: Evidence for a Cell-Autonomous Program of Metabolic Plasticity. Toxins 2025, 17, 293. https://doi.org/10.3390/toxins17060293
dos Santos MP, Leocadio VE, de Sá Hayashide L, Marques M, Carvalho CF, Galina A, Diniz LP. Lipopolysaccharide Induces Mitochondrial Fragmentation and Energetic Shift in Reactive Microglia: Evidence for a Cell-Autonomous Program of Metabolic Plasticity. Toxins. 2025; 17(6):293. https://doi.org/10.3390/toxins17060293
Chicago/Turabian Styledos Santos, Marcelle Pereira, Vitor Emanuel Leocadio, Lívia de Sá Hayashide, Mariana Marques, Clara Fernandes Carvalho, Antonio Galina, and Luan Pereira Diniz. 2025. "Lipopolysaccharide Induces Mitochondrial Fragmentation and Energetic Shift in Reactive Microglia: Evidence for a Cell-Autonomous Program of Metabolic Plasticity" Toxins 17, no. 6: 293. https://doi.org/10.3390/toxins17060293
APA Styledos Santos, M. P., Leocadio, V. E., de Sá Hayashide, L., Marques, M., Carvalho, C. F., Galina, A., & Diniz, L. P. (2025). Lipopolysaccharide Induces Mitochondrial Fragmentation and Energetic Shift in Reactive Microglia: Evidence for a Cell-Autonomous Program of Metabolic Plasticity. Toxins, 17(6), 293. https://doi.org/10.3390/toxins17060293