A Metalloproteinase Cocktail from the Venom of Protobothrops flavoviridis Cleaves Amyloid Beta Peptides at the α-Cleavage Site

 , , ,
, , , 

Abstract
1. Introduction
2. Results
2.1. Fractionation of Snake Venom Metalloproteinases
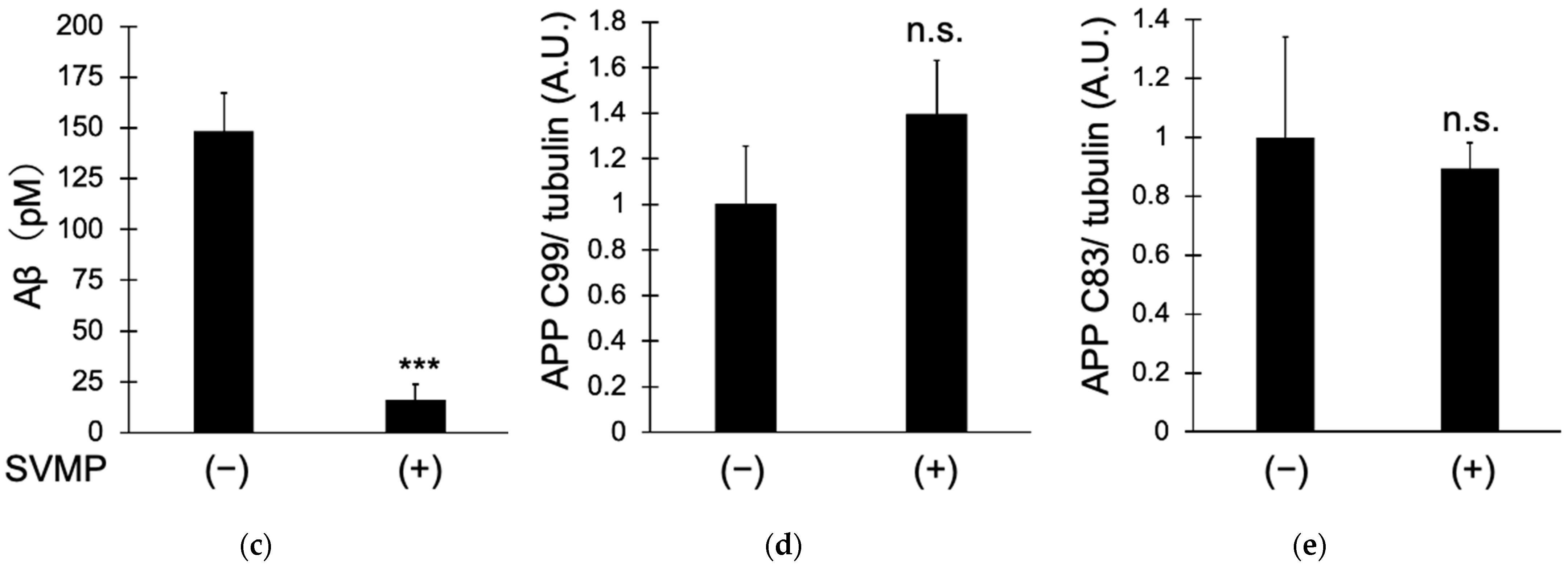
2.2. SVMPs Reduced Aβ Production by Cultured Human Cells
2.3. SVMPs Cleaved Aβ40 and Aβ42 In Vitro at α-Cleavage Sites
2.4. SVMPs Did Not Cleave Thioflavin-Positive Aβ Fibers but Inhibited Their Formation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Fractionation of SVMPs from Crude Venom of P. flavoviridis
4.3. Caseinolytic Assay
4.4. Cytotoxicity Assay
4.5. Aβ Production in NLNTK Cells
4.6. Proteolysis of Aβ40 and Aβ42 Peptides
4.7. Aβ Fibril Formation Detection through ThT Assay
4.8. LC-MS/MS Conditions
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takeda, S. ADAM and ADAMTS family proteins and snake venom metalloproteinases: A structural overview. Toxins 2016, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef]
- Oliveira, A.L.; Viegas, M.F.; Da Silva, S.L.; Soares, A.M.; Ramos, M.J.; Fernandes, P. The chemistry of snake venom and its medical potential. Nat. Rev. Chem. 2022, 6, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Baramova, E.N.; Shannon, J.D.; Bjarnason, J.B.; Gonias, S.L.; Fox, J.W. Interaction of hemorrhagic metalloproteinase with human alpha 2-macroglobulin. Biochemistry 1990, 29, 1069–1074. [Google Scholar] [CrossRef]
- Shibata, H.; Chijiwa, T.; Oda-Ueda, N.; Nakamura, H.; Yamaguchi, K.; Hattori, S.; Matsubara, K.; Matsuda, Y.; Yamashita, A.; Isomoto, A.; et al. The habu genome reveals accelerated evolution of venom protein genes. Sci. Rep. 2018, 8, 11300. [Google Scholar] [CrossRef]
- Ogawa, T.; Oda-Ueda, N.; Hisata, K.; Nakamura, H.; Chijiwa, T.; Hattori, S.; Isomoto, A.; Yugeta, H.; Yamasaki, S.; Fukumaki, Y.; et al. Alternative mRNA splicing in three venom families underlying a possible production of divergent venom proteins of habu snake Protobothrops flavoviridis. Toxins 2019, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R. On the ancestral recruitment of metallo proteinases int othe venom of snakes. Toxicon 2012, 60, 449–454. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Strooper, D.; Vassar, R.; Golde, T. The secretase: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Szaruga, M.; Veugelen, S.; Benurwar, M.; Lismont, S.; Sepulveda-Falla, D.; Lleo, A.; Ryan, N.S.; Lashley, T.; Fox, N.C.; Murayama, S.; et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J. Exp. Med. 2015, 212, 2003–2013. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. A close look at BACE1 inhibitors for Alzheimer’s disease treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T. Molecular mechanism of intramembrane proteolysis by γ-secretase. J. Biochem. 2014, 156, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Frisardi, V.; Capurso, C.; D’Introno, A.; Colacicco, A.M.; Vendamiale, G.; Capurso, A.; Imbimbo, B.P. Disease-modifying approach to the treatment of Alzheimer’s disease. Drugs Aging 2009, 26, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Athar, T.; Balushi, K.; Khan, S.A. Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 5629–5645. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Drug Approval Package: Aduhelm (Aducanumab-Avwa). Office of Neurology’s Summary Review Memorandum. FDA. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000TOC.cfm (accessed on 6 August 2023).
- Karron, E.; De Strooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug. Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Cushman, D.W.; Cheung, H.S.; Sabo, E.F.; Ondetti, M.A. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 1977, 16, 5484–5491. [Google Scholar] [CrossRef]
- Hartman, G.D.; Egbertson, M.S.; Halczenko, W.; Laswell, W.L.; Duggan, M.E.; Smith, R.L.; Naylor, A.M.; Manno, P.D.; Lynch, R.J.; Zhang, G.; et al. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992, 35, 4640–4642. [Google Scholar] [CrossRef]
- Scarborough, R.M. Development of eptibatide. Am. Heart J. 1999, 138, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Sato, A.; Shizuno, T.; Yokoyama, K.; Suzuki, Y.; Tokunaga, M.; Asahara, T. Batroxobin accelerated tissue repair via neutrophil extracellular trap regulation and defibrinogenation in a murine ischemic hind limd model. PLoS ONE 2019, 14, e0220898. [Google Scholar] [CrossRef] [PubMed]
- Bordon, K.C.F.; Cologna, C.T.; Fornari-Baldo, E.C.; Pinheiro-Júnior, E.L.; Cerni, F.A.; Amorim, F.G.; Anjolette, F.A.P.; Cordeiro, F.A.; Wiezel, G.A.; Cardoso, I.A.; et al. From animal poisons and venoms to medicine: Achievements, challenges and perspectives in drug discovery. Front. Pharmacol. 2020, 11, 1132. [Google Scholar] [CrossRef]
- Wijeyewickrema, L.C.; Gardiner, E.E.; Shen, Y.; Berndt, M.C.; Andrews, R.K. Fractionation of snake venom metallo proteinase by metal ion affinity: A purified cobra metalloproteinase, Nk, from Naja kaouthia binds Ni2+-agarose. Toxicon 2007, 50, 1064–1072. [Google Scholar] [CrossRef]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef]
- Kikuchi, K.; Kidana, K.; Tatebe, T.; Tomita, T. Dysregulated metabolism of the amyloid β-protein and therapeutic approaches in Alzheimer disease. J. Cell. Biochem. 2017, 118, 4183–4190. [Google Scholar] [CrossRef] [PubMed]
- Kidana, K.; Tatebe, T.; Ito, K.; Hara, N.; Kakita, A.; Saito, T.; Takatori, S.; Ouchi, Y.; Ikeuchi, T.; Makino, M.; et al. Loss of kallikrein-related peptidase 7 exacerbates amyloid pathology in Alzheimer’s disease model mice. EMBO Mol. Med. 2018, 10, e8184. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Sekiguchi, M.; Hattori, Y.; Takahashi, A.; Asai, M.; Ji, B.; Higuchi, M.; Staufenbiel, M.; Muramatsu, S.; Saido, T.C. Global brain delivery of neprilysin gene by intravascular administration of AAV vector in mice. Sci. Rep. 2013, 3, 1472. [Google Scholar] [CrossRef]
- Saito, T.; Iwata, N.; Tsubuki, S.; Takaki, Y.; Takano, J.; Huang, S.M.; Suemoto, T.; Higuchi, M.; Saido, T.C. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat. Med. 2005, 11, 434–439. [Google Scholar] [CrossRef]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef]
- Malmberg, A.B.; Yaksh, T.L. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995, 60, 83–90. [Google Scholar] [CrossRef]
- Liao, M.C.; Ahmed, M.; Smith, S.O.; Van Nostrand, W.E. Degradation of amyloid β protein by purified myelin basic protein. J. Biol. Chem. 2009, 284, 28917–28925. [Google Scholar] [CrossRef]
- Tucher, J.; Linke, D.; Koudelka, T.; Cassidy, L.; Tredup, C.; Wichert, R.; Pietzik, C.; Becker-pauly, C.; Tholey, A. LC-MS based cleavagesite profiling of the proteases ADAM10 and ADAM17 using proteome derived peptide libraries. J. Proteome. Res. 2014, 13, 2205–2214. [Google Scholar] [CrossRef]
- Seo, T.; Sakon, T.; Nakazawa, S.; Nishioka, A.; Watanabe, K.; Matsumoto, K.; Akasaka, M.; Shioi, N.; Sawada, H.; Araki, S. Haemorragic snake venom metalloproteases and human ADAMs cleave LRP5/6, which disrupts cell-cell adhesions in vitro and induces haemorrhage in vivo. FEBS J. 2017, 284, 1657–1671. [Google Scholar] [CrossRef]
- Hotoda, N.; Koike, H.; Sasagawa, N.; Ishiura, S. A secreted form of human ADAM9 has an α-secretase activity for APP. Biohem. Biophys. Res. Commun. 2002, 293, 800–805. [Google Scholar] [CrossRef]
- Kurosumi, M.; Nishio, Y.; Osawa, S.; Kobayashi, H.; Iwatsubo, T.; Tomita, T.; Miyachi, H. Novel Notch-sparing γ-secretase inhibitors derived from a peroxisome proliferator-activated receptor agonist library. Bioorg. Med. Chem. Lett. 2010, 20, 5282–5285. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Kihara, H.; Ohno, M. Purification and properties of phospholipase A from venom of Trimeresurus flavoviridis (Habu Snake). J. Biochem. 1980, 88, 443–451. [Google Scholar] [CrossRef]
- Ishiura, S.; Murofushi, H.; Suzuki, K.; Imahori, K. Studies of calcium-activated neutral protease from chicken skeletal muscle. I. Purification and characterization. J. Biochem. 1978, 84, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, S.; Eugene, F.; Ishiura, S. In vitro reconstitution of γ-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 2008, 377, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hashimoto, T.; Nomoto, H.; Hyman, B.T.; Iwatsubo, T. Role of apolipoprotein E in β-amyloidogenesis: Isoform-specific effects on protofibril to fibril conversion of Aβ in vitro and brain Aβ deposition in vivo. J. Biol. Chem. 2015, 290, 15163–15174. [Google Scholar] [CrossRef]
- Ogawa, T.; Tobishima, Y.; Kamata, S.; Matsuda, Y.; Muramoto, K.; Hidaka, M.; Eugene, F.; Kuraishi, T.; Yokota, S.; Ohno, M.; et al. Focused proteomics analysis of Habu snake (Protobothrops flavoviridis) venom using antivenom-based affinity chromatography reveals novel myonecrosis-enhancing activity of thrombin-like serine proteases. Front. Pharmacol. 2021, 12, 766406. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | SVMP Class | Molecular Mass * (Da) | Length * (Amino Acids) | Sequence Coverage * (%) | Intensity |
|---|---|---|---|---|---|
| svMP08_flavoridin | P-II | 54,503 | 483 | 27.5 | 5,524,700 |
| svMP06_H2pro | P-I | 46,405 | 408 | 28.7 | 4,375,700 |
| svMP09_HR1b | P-III | 69,129 | 615 | 19.7 | 1,074,000 |
| svMP07_HR2a | P-II | 53,991 | 480 | 35.8 | 903,290 |
| svMP05_HR1a | P-III | 68,765 | 609 | 6.9 | 518,200 |
| svMP02_flavorase | P-III | 69,015 | 614 | 8.0 | 125,290 |
| svMP03_VMP-III_like | P-III | 69,657 | 620 | 8.1 | 70,111 |
| svMP01_HV1 | P-III | 68,176 | 612 | 11.1 | 37,075 |
| svMP04_jerdonitin_like | P-II | 53,872 | 480 | 6.5 | 13,726 |
| Sequence | Charges | m/z | Intensity | Ratio |
|---|---|---|---|---|
| DAEFRHDSGYEVHHQK | 3 | 652.30 | 320,540 | 1.0 |
| DAEFRHDSGYEVHHQKLV | 4 | 542.51 | 14,529 | 0.045 |
| DAEFRHDSGYEVHHQKLVFFAEDVGSNKGA | 5 | 678.72 | 694.87 | 0.0022 |
| IIGLMVGGVV | 2 | 479.29 | 499.04 | 0.0016 |
| Sequence | Charges | m/z | Intensity | Ratio |
|---|---|---|---|---|
| DAEFRHDSGYEVHHQK | 3 | 652.3 | 428,430 | 1.0 |
| DAEFRHDSGYEVHHQKLVFFAEDVGSNKGA | 5 | 678.22 | 98,285 | 0.23 |
| DAEFRHDSGYEVHHQKLV | 4 | 542.51 | 4817.5 | 0.011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Futai, E.; Kawasaki, H.; Sato, S.; Daoudi, K.; Hidaka, M.; Tomita, T.; Ogawa, T. A Metalloproteinase Cocktail from the Venom of Protobothrops flavoviridis Cleaves Amyloid Beta Peptides at the α-Cleavage Site. Toxins 2023, 15, 500. https://doi.org/10.3390/toxins15080500
Futai E, Kawasaki H, Sato S, Daoudi K, Hidaka M, Tomita T, Ogawa T. A Metalloproteinase Cocktail from the Venom of Protobothrops flavoviridis Cleaves Amyloid Beta Peptides at the α-Cleavage Site. Toxins. 2023; 15(8):500. https://doi.org/10.3390/toxins15080500
Chicago/Turabian StyleFutai, Eugene, Hajime Kawasaki, Shinichi Sato, Khadija Daoudi, Masafumi Hidaka, Taisuke Tomita, and Tomohisa Ogawa. 2023. "A Metalloproteinase Cocktail from the Venom of Protobothrops flavoviridis Cleaves Amyloid Beta Peptides at the α-Cleavage Site" Toxins 15, no. 8: 500. https://doi.org/10.3390/toxins15080500
APA StyleFutai, E., Kawasaki, H., Sato, S., Daoudi, K., Hidaka, M., Tomita, T., & Ogawa, T. (2023). A Metalloproteinase Cocktail from the Venom of Protobothrops flavoviridis Cleaves Amyloid Beta Peptides at the α-Cleavage Site. Toxins, 15(8), 500. https://doi.org/10.3390/toxins15080500