The Functions of Cholera Toxin Subunit B as a Modulator of Silica Nanoparticle Endocytosis

 ,
,  , ,
, ,  and
and
Abstract
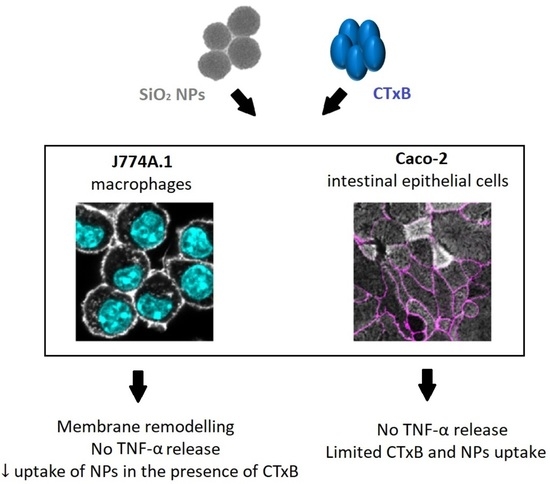
1. Introduction
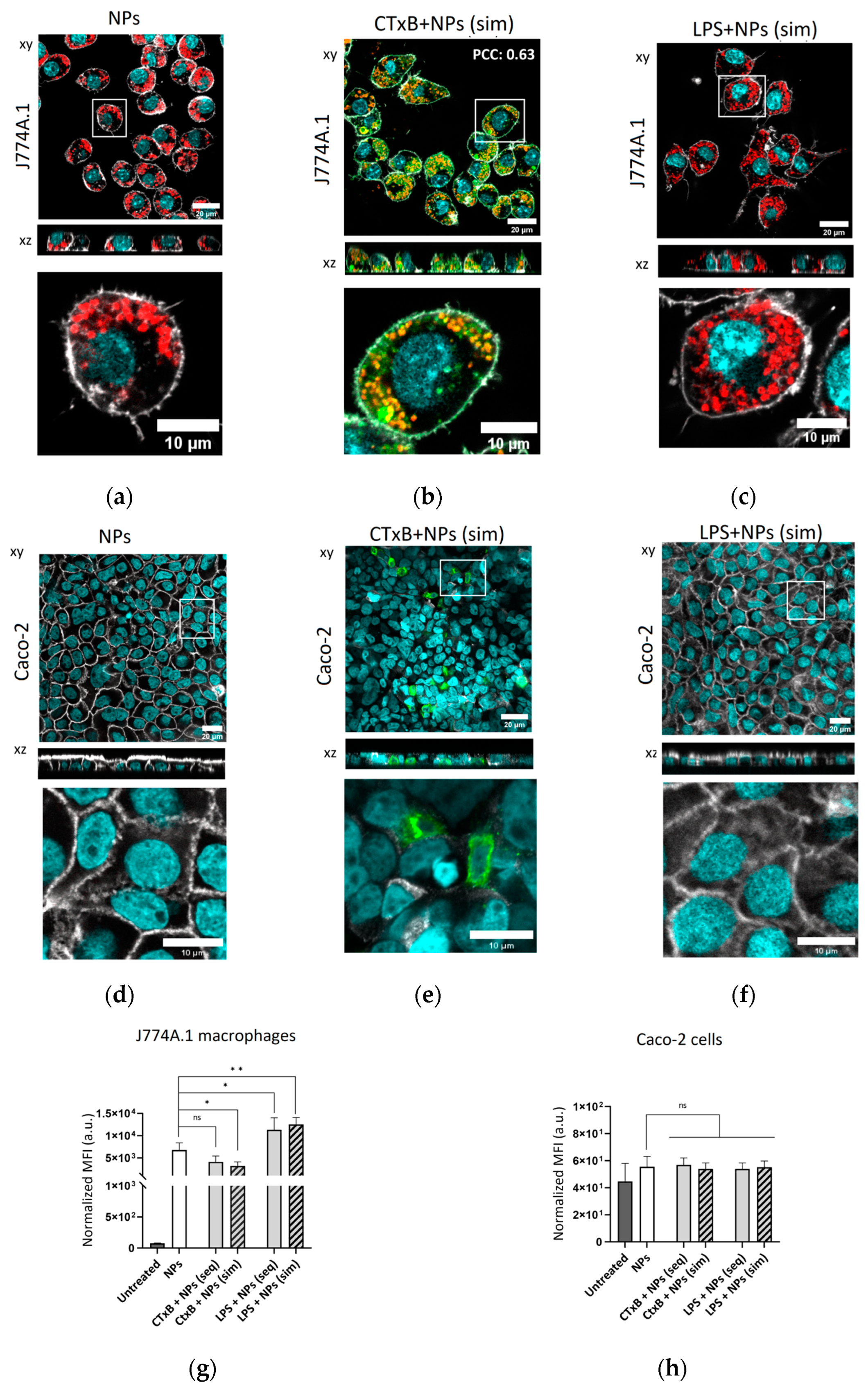
2. Results and Discussion
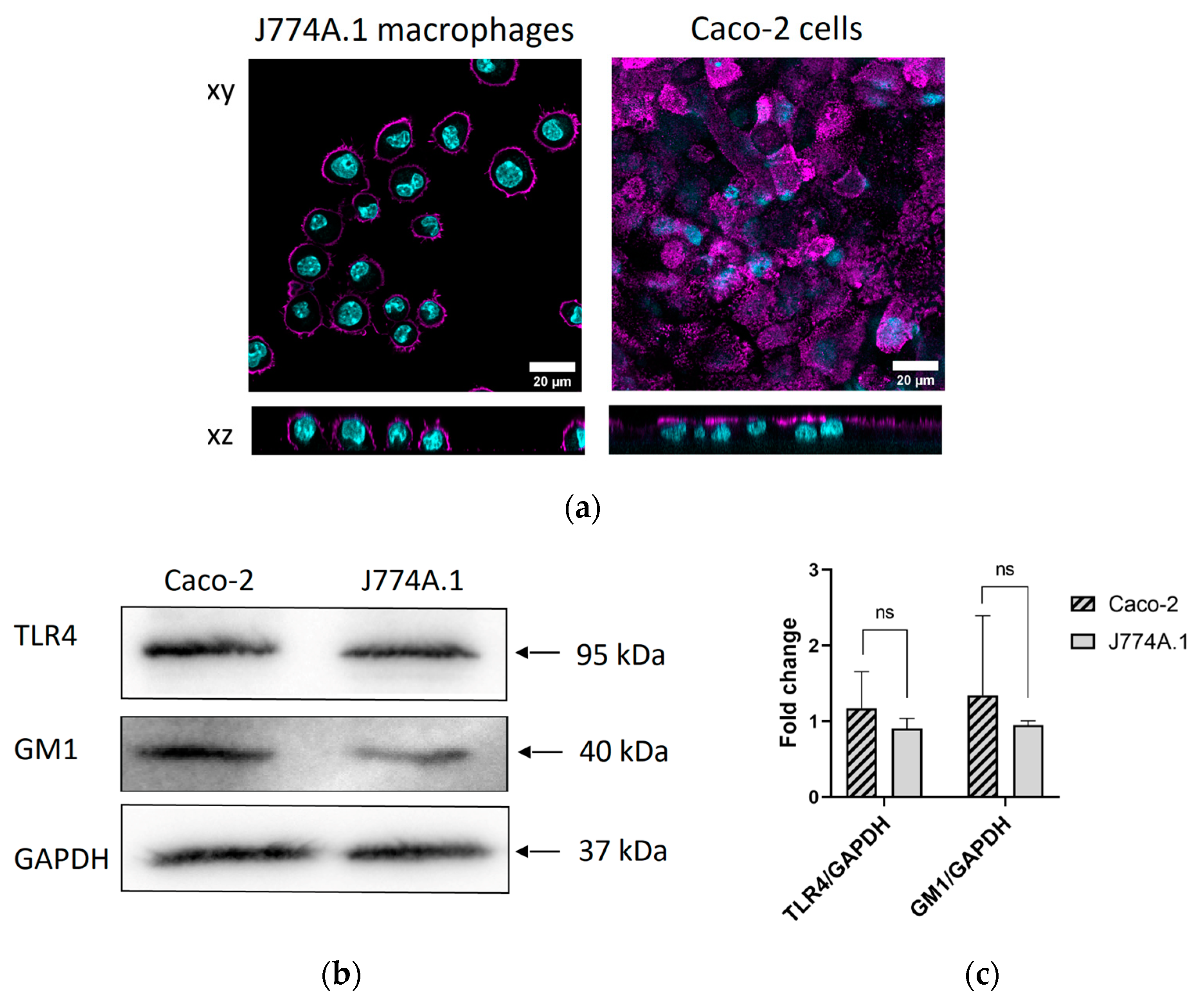
2.1. Surface Expression of GM1 and TLR4 Receptors
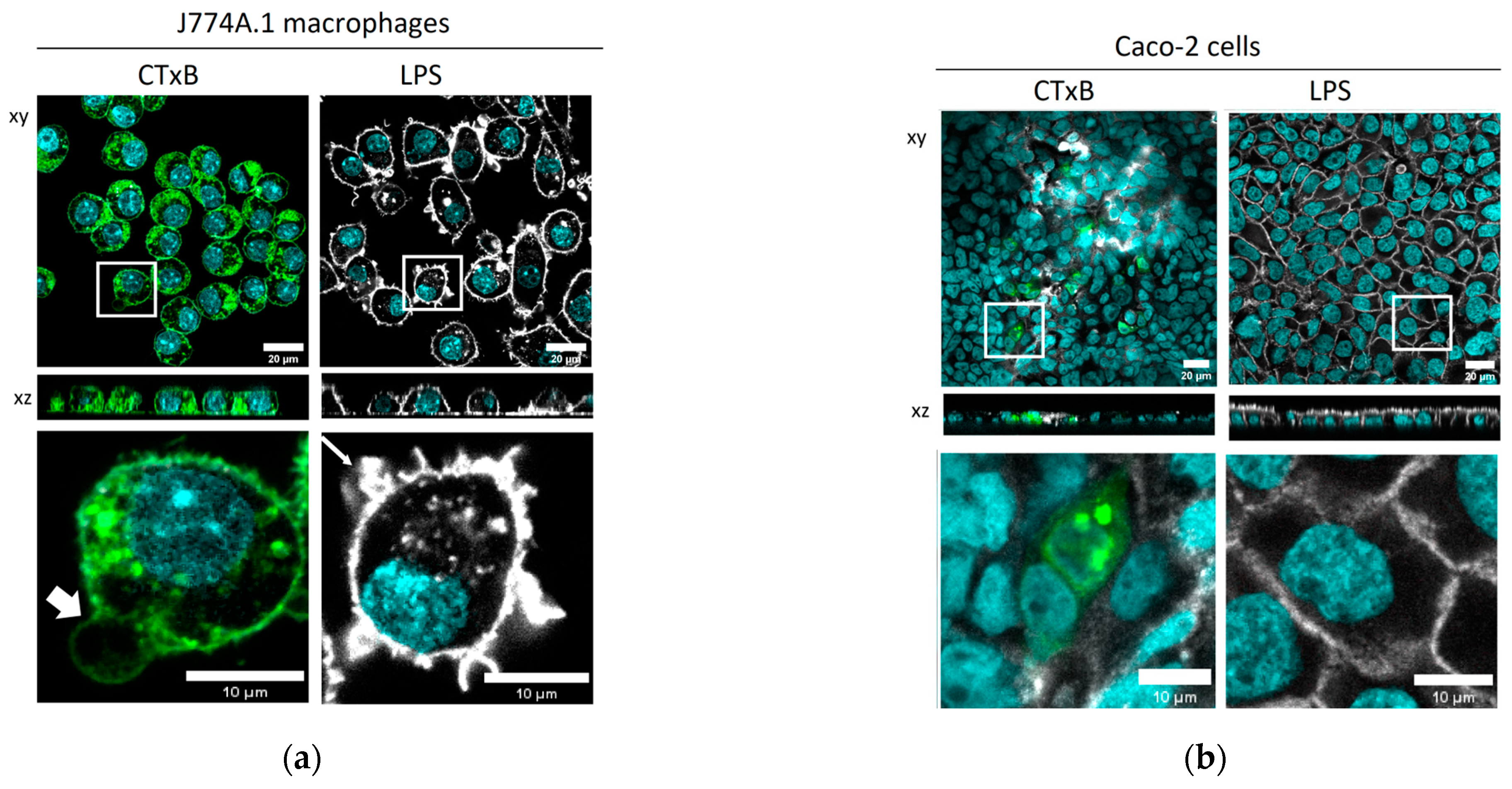
2.2. The Cellular Morphology of Macrophages Is Altered by CTxB Treatment
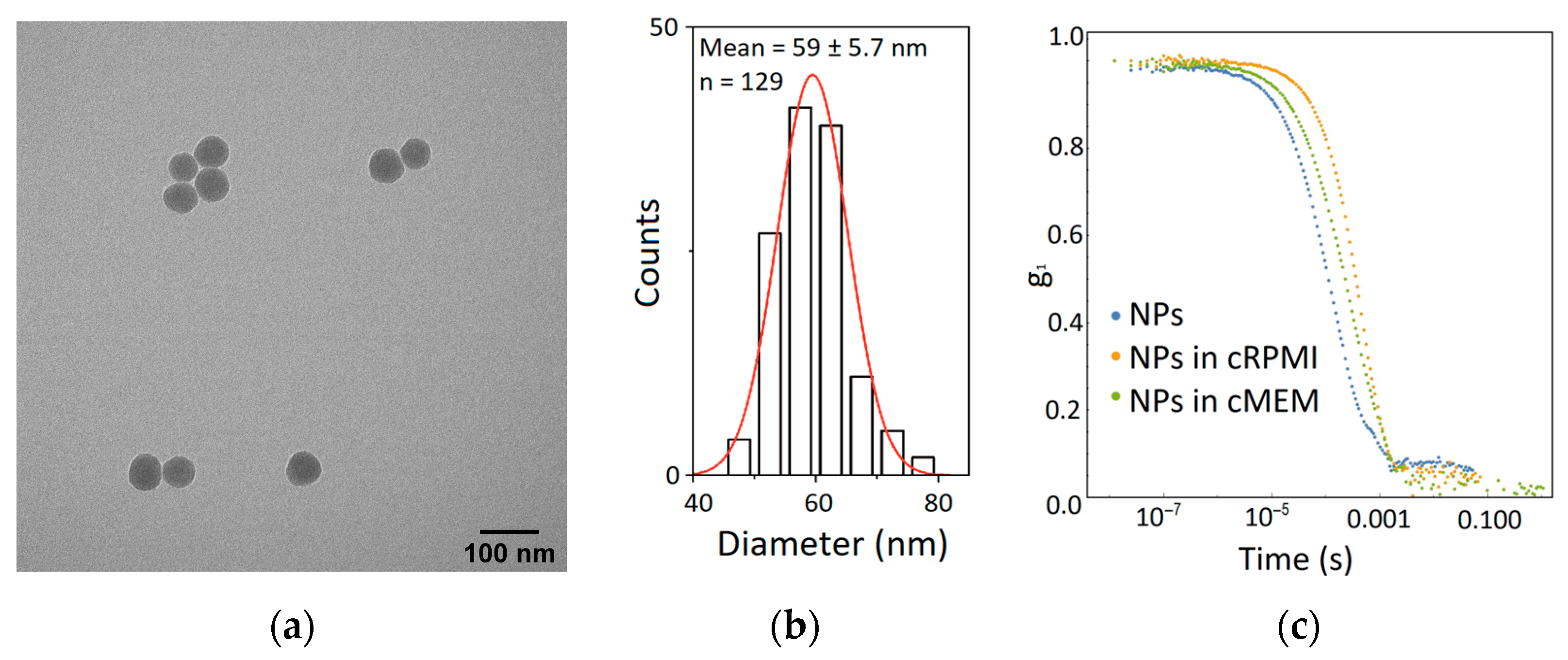
2.3. Nanoparticle Characterization
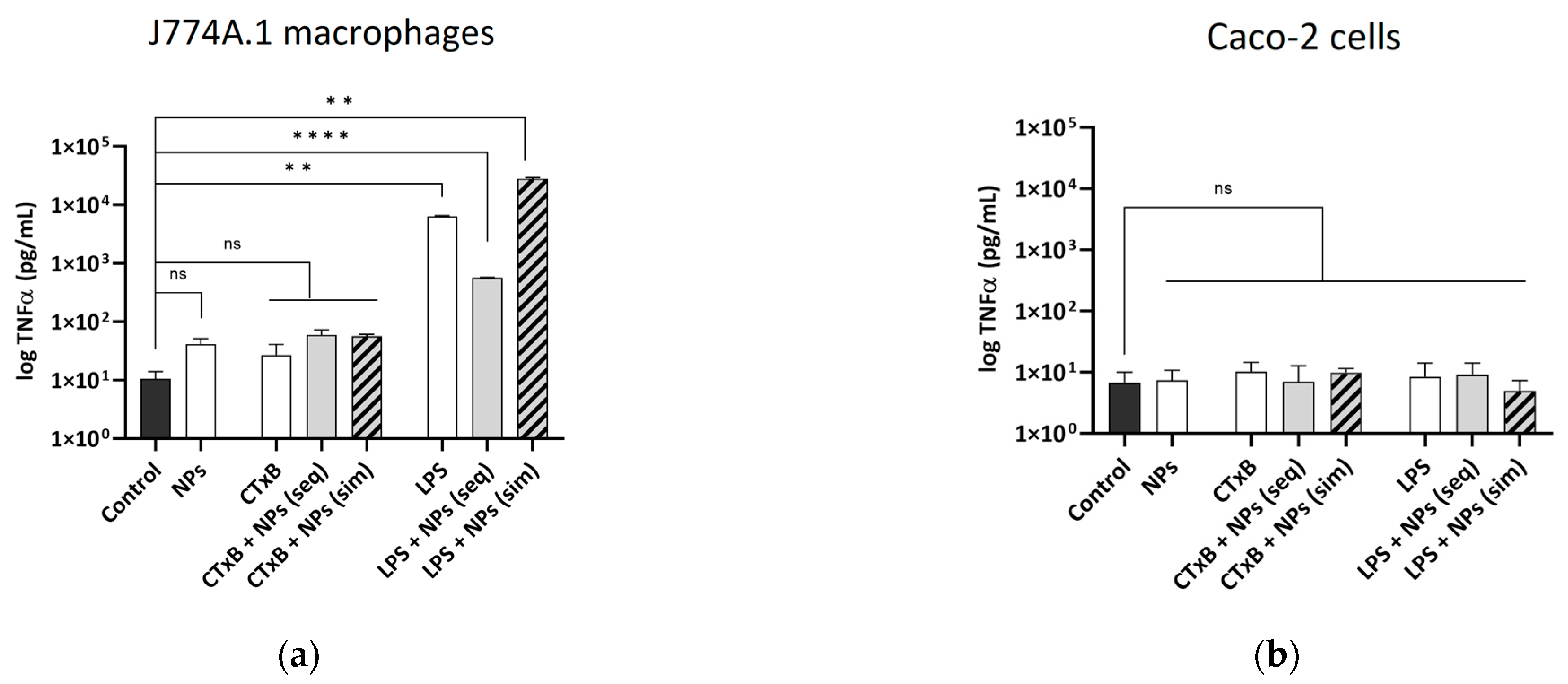
2.4. The Effect of CTxB on the Secretion of Pro-Inflammatory Cytokine TNF-α
2.5. The Effect of CTxB on the Cellular Association and Uptake of Silica NPs
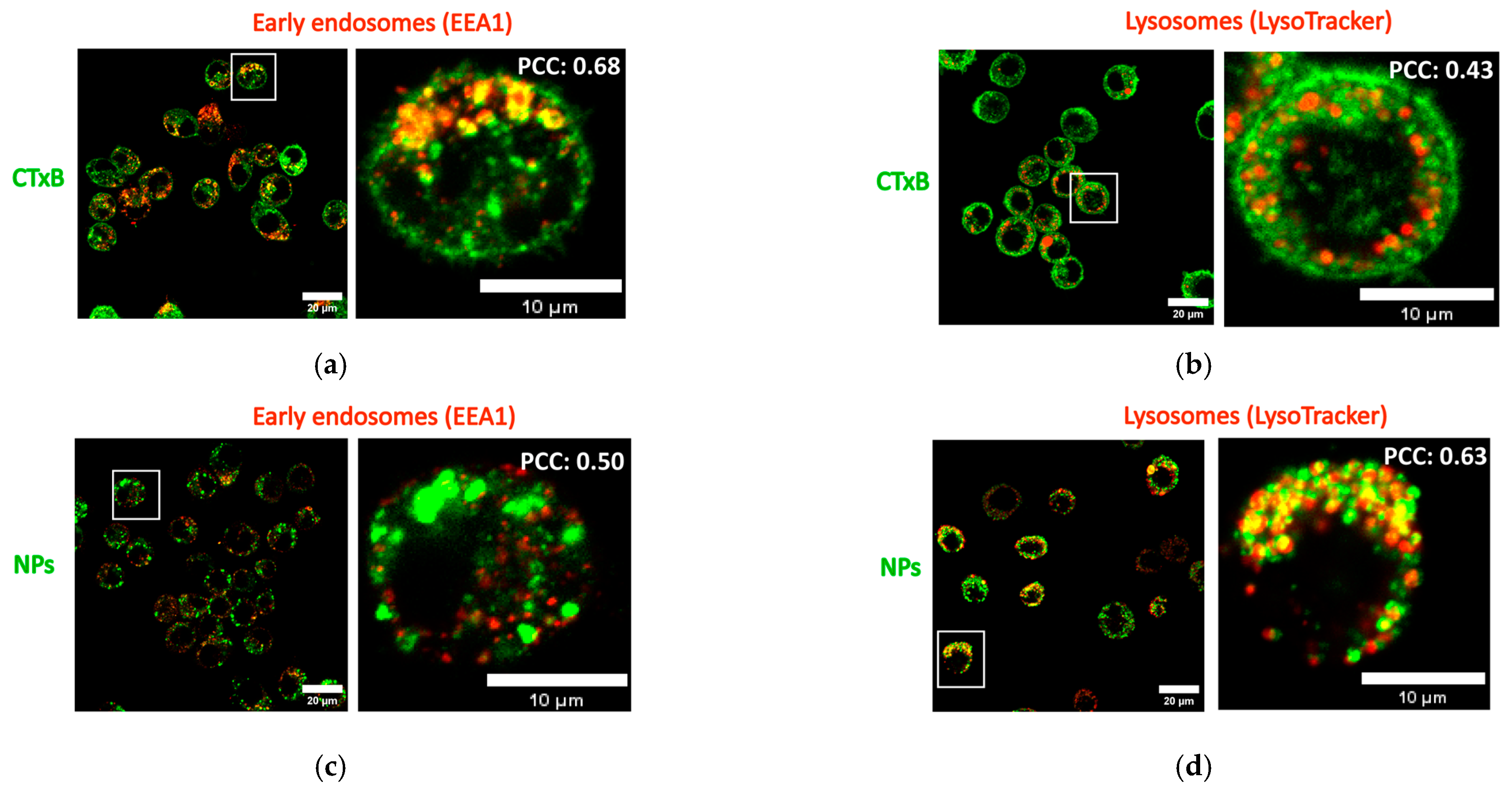
2.6. Intracellular Localization of NPs in Macrophages in the Presence of CTxB
3. Conclusions
4. Materials and Methods
4.1. Nanoparticle Synthesis and Characterization
4.2. Dye Leaching from SiO2 NPs
4.3. Cell Culture
4.4. Western Blot
4.5. Cell Permeability
4.6. Cell Exposures
- Single: The cells were exposed to 59 nm SiO2-BDP FL NPs dispersed in cRPMI/cMEM for 24 h.
- Sequential: The cells were initially pre-treated with either 1 µg/mL CTxB (C34778, Thermo Fisher Scientific) or 1 µg/mL LPS (Escherichia coli strain O111:B4, Cat. No. L4391, Sigma-Aldrich) in complete RPMI medium (cRPMI) or complete MEM (cMEM) for a duration of 24h. After the pre-treatment period, the CTxB or LPS was removed, and the cells were subsequently exposed to NPs dispersed in cRPMI or cMEM for an additional 24 h.
- Simultaneous: A combination of NPs (20 µg/mL) and CTxB (1 µg/mL) or LPS (1 µg/mL) was pre-mixed in cRPMI or cMEM and applied to the cells for 24 h.
4.7. Cell Viability
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Immunofluorescence and Confocal Microscopy
4.10. Colocalization Analysis
4.11. Flow Cytometry
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, G.-H.; Kim, Y.-S.; Kwon, E.; Yun, J.-W.; Kang, B.-C. Toxicologic Evaluation for Amorphous Silica Nanoparticles: Genotoxic and Non-Genotoxic Tumor-Promoting Potential. Pharmaceutics 2020, 12, 826. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.M.; Cordeiro, R.A.; Faneca, H. Silica-Based Gene Delivery Systems: From Design to Therapeutic Applications. Pharmaceutics 2020, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, P.; Zhao, R.; Zhao, L.; Liu, J.; Peng, S.; Fu, X.; Wang, X.; Luo, R.; Wang, R.; et al. Silica nanoparticles: Biomedical applications and toxicity. Biomed. Pharmacother. 2022, 151, 113053. [Google Scholar] [CrossRef] [PubMed]
- Contado, C.; Mejia, J.; Lozano García, O.; Piret, J.-P.; Dumortier, E.; Toussaint, O.; Lucas, S. Physicochemical and toxicological evaluation of silica nanoparticles suitable for food and consumer products collected by following the EC recommendation. Anal. Bioanal. Chem. 2016, 408, 271–286. [Google Scholar] [CrossRef]
- Bourquin, J.; Milosevic, A.; Hauser, D.; Lehner, R.; Blank, F.; Petri-Fink, A.; Rothen-Rutishauser, B. Biodistribution, Clearance, and Long-Term Fate of Clinically Relevant Nanomaterials. Adv. Mater. 2018, 30, e1704307. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.M. Gut bacteria in health and disease. Gastroenterol. Hepatol. 2013, 9, 560–569. [Google Scholar]
- Sánchez, J.; Holmgren, J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. 2008, 65, 1347–1360. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera toxin subunit b as adjuvant—An accelerator in protective immunity and a break in autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Skretting, G.; Van Deurs, B.; Sandvig, K. Internalization of cholera toxin by different endocytic mechanisms. J. Cell Sci. 2001, 114, 3737–3747. [Google Scholar] [CrossRef]
- Rodighiero, C.; Fujinaga, Y.; Hirst, T.R.; Lencer, W.I. A Cholera Toxin B-subunit Variant that Binds Ganglioside GM1 but Fails to Induce Toxicity. J. Biol. Chem. 2001, 276, 36939–36945. [Google Scholar] [CrossRef]
- Baldauf, K.J.; Royal, J.M.; Hamorsky, K.T.; Matoba, N. Cholera toxin B: One subunit with many pharmaceutical applications. Toxins 2015, 7, 974–996. [Google Scholar] [CrossRef]
- Schnitzler, A.C.; Burke, J.M.; Wetzler, L.M. Induction of cell signaling events by the cholera toxin B subunit in antigen-presenting cells. Infect. Immun. 2007, 75, 3150–3159. [Google Scholar] [CrossRef]
- Phongsisay, V.; Iizasa, E.; Hara, H.; Yoshida, H. Evidence for TLR4 and FcRγ-CARD9 activation by cholera toxin B subunit and its direct bindings to TREM2 and LMIR5 receptors. Mol. Immunol. 2015, 66, 463–471. [Google Scholar] [CrossRef]
- Orimo, T.; Sasaki, I.; Hemmi, H.; Ozasa, T.; Fukuda-Ohta, Y.; Ohta, T.; Morinaka, M.; Kitauchi, M.; Yamaguchi, T.; Sato, Y.; et al. Cholera toxin B induces interleukin-1β production from resident peritoneal macrophages through the pyrin inflammasome as well as the NLRP3 inflammasome. Int. Immunol. 2019, 31, 657–668. [Google Scholar] [CrossRef]
- Burkart, V.; Kim, Y.-E.; Hartmann, B.; Ghiea, I.; Syldath, U.; Kauer, M.; Fingberg, W.; Hanifi-Moghaddam, P.; Müller, S.; Kolb, H. Cholera Toxin B Pretreatment of Macrophages and Monocytes Diminishes Their Proinflammatory Responsiveness to Lipopolysaccharide. J. Immunol. 2002, 168, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Li, T.K.; Fox, B.S. Cholera toxin B subunit binding to an antigen-presenting cell directly costimulates cytokine production from a T cell clone. Int. Immunol. 1996, 8, 1849–1856. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soo, J.-C.; Zhang, J.; He, Q.; Agarwal, S.; Li, H.; Zhang, H.; Chen, P. Surface immobilized cholera toxin B subunit (CTB) facilitates vesicle docking, trafficking and exocytosis. Integr. Biol. 2010, 2, 250. [Google Scholar] [CrossRef]
- Susnik, E.; Taladriz-Blanco, P.; Drasler, B.; Balog, S.; Petri-Fink, A.; Rothen-Rutishauser, B. Increased Uptake of Silica Nanoparticles in Inflamed Macrophages but Not upon Co-Exposure to Micron-Sized Particles. Cells 2020, 9, 2099. [Google Scholar] [CrossRef]
- Walker, W.A.; Tarannum, M.; Vivero-Escoto, J.L. Cellular endocytosis and trafficking of cholera toxin B-modified mesoporous silica nanoparticles. J. Mater. Chem. B 2016, 4, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Wiedinger, K.; Romlein, H.; Bitsaktsis, C. Cholera toxin B induced activation of murine macrophages exposed to a fixed bacterial immunogen. Ther. Adv. Vaccines 2015, 3, 155–163. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Bramini, M.; Hristov, D.R.; Wan, S.; Salvati, A.; Åberg, C.; Dawson, K.A. Low uptake of silica nanoparticles in Caco-2 intestinal epithelial barriers. Beilstein J. Nanotechnol. 2017, 8, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Jahn, K.A.; Biazik, J.M.; Braet, F. GM1 Expression in Caco-2 Cells: Characterisation of a Fundamental Passage-dependent Transformation of a Cell Line. J. Pharm. Sci. 2011, 100, 3751–3762. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.A.; Vanhecke, D.; Michen, B.; Blank, F.; Gehr, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 2014, 5, 1625–1636. [Google Scholar] [CrossRef]
- Pang, H.; Le, P.U.; Nabi, I.R. Ganglioside GM1 levels are a determinant of the extent of caveolae/raft-dependent endocytosis of cholera toxin to the Golgi apparatus. J. Cell Sci. 2004, 117, 1421–1430. [Google Scholar] [CrossRef]
- Groza, R.; Ewers, H. Membrane deformation by the cholera toxin beta subunit requires more than one binding site. Proc. Natl. Acad. Sci. USA 2020, 117, 17467–17469. [Google Scholar] [CrossRef]
- Kabbani, A.M.; Kelly, C.V. Nanoscale Membrane Budding Induced by CTxB and Detected via Polarized Localization Microscopy. Biophys. J. 2017, 113, 1795–1806. [Google Scholar] [CrossRef]
- Kim, E.J.; Lee, M.Y.; Jeon, Y.J. Silymarin Inhibits Morphological Changes in LPS-Stimulated Macrophages by Blocking NF-κB Pathway. Korean J. Physiol. Pharmacol. 2015, 19, 211. [Google Scholar] [CrossRef]
- Masarudin, M.J.; Cutts, S.M.; Evison, B.J.; Phillips, D.R.; Pigram, P.J. Factors determining the stability, size distribution, and cellular accumulation of small, monodisperse chitosan nanoparticles as candidate vectors for anticancer drug delivery: Application to the passive encapsulation of [14C]-doxorubicin. Nanotechnol. Sci. Appl. 2015, 67, 67–80. [Google Scholar] [CrossRef]
- Mohr, K.; Müller, S.S.; Müller, L.K.; Rusitzka, K.; Gietzen, S.; Frey, H.; Schmidt, M. Evaluation of Multifunctional Liposomes in Human Blood Serum by Light Scattering. Langmuir 2014, 30, 14954–14962. [Google Scholar] [CrossRef]
- Crippa, F.; Rodriguez-Lorenzo, L.; Hua, X.; Goris, B.; Bals, S.; Garitaonandia, J.S.; Balog, S.; Burnand, D.; Hirt, A.M.; Haeni, L.; et al. Phase Transformation of Superparamagnetic Iron Oxide Nanoparticles via Thermal Annealing: Implications for Hyperthermia Applications. ACS Appl. Nano Mater. 2019, 2, 4462–4470. [Google Scholar] [CrossRef]
- Baldauf, K.J.; Royal, J.M.; Kouokam, J.C.; Haribabu, B.; Jala, V.R.; Yaddanapudi, K.; Hamorsky, K.T.; Dryden, G.W.; Matoba, N. Oral administration of a recombinant cholera toxin B subunit promotes mucosal healing in the colon. Mucosal Immunol. 2017, 10, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhaya, A.; Sarkar, M.; Chaudhuri, K. Transcriptional upregulation of inflammatory cytokines in human intestinal epithelial cells following Vibrio cholerae infection. FEBS J. 2007, 274, 4631–4642. [Google Scholar] [CrossRef] [PubMed]
- Ude, V.C.; Brown, D.M.; Viale, L.; Kanase, N.; Stone, V.; Johnston, H.J. Impact of copper oxide nanomaterials on differentiated and undifferentiated Caco-2 intestinal epithelial cells; assessment of cytotoxicity, barrier integrity, cytokine production and nanomaterial penetration. Part. Fibre Toxicol. 2017, 14, 31. [Google Scholar] [CrossRef]
- Day, C.A.; Kenworthy, A.K. Functions of cholera toxin B-subunit as a raft cross-linker. Essays Biochem. 2015, 57, 135–145. [Google Scholar] [CrossRef]
- Rouquette-Jazdanian, A.K.; Foussat, A.; Lamy, L.; Pelassy, C.; Lagadec, P.; Breittmayer, J.-P.; Aussel, C. Cholera Toxin B-Subunit Prevents Activation and Proliferation of Human CD4 + T Cells by Activation of a Neutral Sphingomyelinase in Lipid Rafts. J. Immunol. 2005, 175, 5637–5648. [Google Scholar] [CrossRef]
- Annika Mareike Gramatke, I.-L.H. Size and Cell Type Dependent Uptake of Silica Nanoparticles. J. Nanomed. Nanotechnol. 2015, 5, 6. [Google Scholar] [CrossRef]
- Iversen, T.G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef]
- Gonzalez Porras, M.A.; Durfee, P.; Giambini, S.; Sieck, G.C.; Brinker, C.J.; Mantilla, C.B. Uptake and intracellular fate of cholera toxin subunit b-modified mesoporous silica nanoparticle-supported lipid bilayers (aka protocells) in motoneurons. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 661–672. [Google Scholar] [CrossRef]
- Moreno-Echeverri, A.M.; Susnik, E.; Vanhecke, D.; Taladriz-Blanco, P.; Balog, S.; Petri-Fink, A.; Rothen-Rutishauser, B. Pitfalls in methods to study colocalization of nanoparticles in mouse macrophage lysosomes. J. Nanobiotechnol. 2022, 18, 464. [Google Scholar] [CrossRef]
- Stober, W. Controlled Growth of Monodisperse Silica Spheres in the Micron Size Range. J. Colloid Interface Sci. 1968, 69, 62–69. [Google Scholar] [CrossRef]
- Moore, T.L.; Urban, D.A.; Rodriguez-Lorenzo, L.; Milosevic, A.; Crippa, F.; Spuch-Calvar, M.; Balog, S.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle administration method in cell culture alters particle-cell interaction. Sci. Rep. 2019, 9, 900. [Google Scholar] [CrossRef]
- Aaron, J.S.; Taylor, A.B.; Chew, T.-L. Image co-localization–co-occurrence versus correlation. J. Cell Sci. 2018, 131, jcs211847. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | NPs | CTxB + NPs | LPS + NPs |
|---|---|---|---|
| Milli-Q water | 85 ± 1 nm | 83 ± 2 nm | 82 ± 2 nm |
| cRPMI | 95 ± 2 nm | 96 ± 2 nm | 98 ± 3 nm |
| cMEM | 94 ± 2 nm | 97 ± 2 nm | 87 ± 1 nm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Susnik, E.; Balog, S.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. The Functions of Cholera Toxin Subunit B as a Modulator of Silica Nanoparticle Endocytosis. Toxins 2023, 15, 482. https://doi.org/10.3390/toxins15080482
Susnik E, Balog S, Taladriz-Blanco P, Petri-Fink A, Rothen-Rutishauser B. The Functions of Cholera Toxin Subunit B as a Modulator of Silica Nanoparticle Endocytosis. Toxins. 2023; 15(8):482. https://doi.org/10.3390/toxins15080482
Chicago/Turabian StyleSusnik, Eva, Sandor Balog, Patricia Taladriz-Blanco, Alke Petri-Fink, and Barbara Rothen-Rutishauser. 2023. "The Functions of Cholera Toxin Subunit B as a Modulator of Silica Nanoparticle Endocytosis" Toxins 15, no. 8: 482. https://doi.org/10.3390/toxins15080482
APA StyleSusnik, E., Balog, S., Taladriz-Blanco, P., Petri-Fink, A., & Rothen-Rutishauser, B. (2023). The Functions of Cholera Toxin Subunit B as a Modulator of Silica Nanoparticle Endocytosis. Toxins, 15(8), 482. https://doi.org/10.3390/toxins15080482