Emodin, an Emerging Mycotoxin, Induces Endoplasmic Reticulum Stress-Related Hepatotoxicity through IRE1α–XBP1 Axis in HepG2 Cells


Abstract
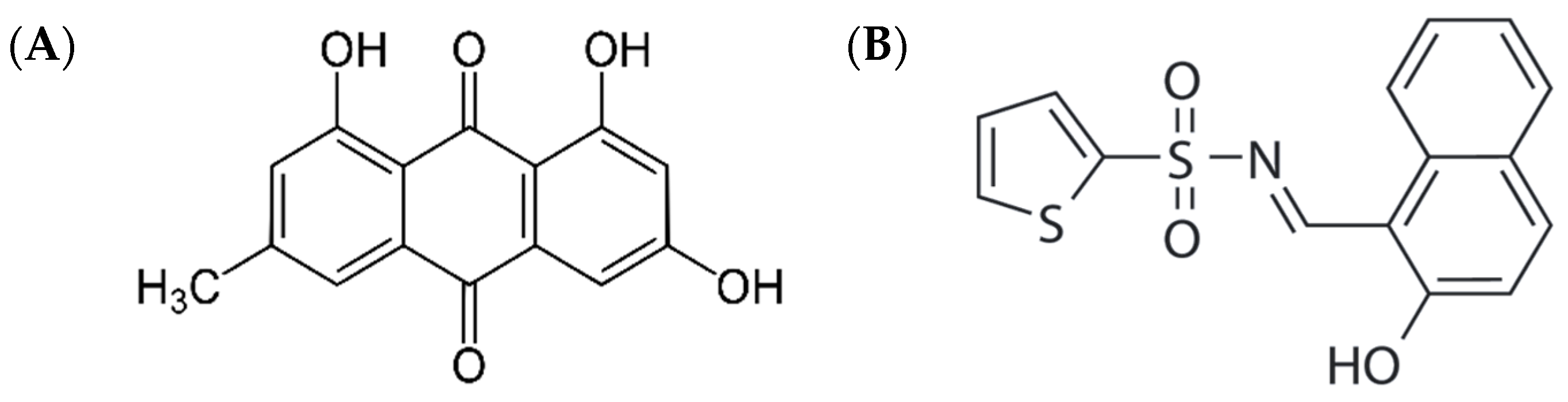
1. Introduction
2. Results
2.1. Decrease in the Metabolic Activity of HepG2 Cells Induced with Emodin
2.2. Changes in Morphological Properties of HepG2 Cells Induced with Emodin
2.3. Changes in the Gene and Protein Expression of ER Stress and Apoptosis Markers Induced with Emodin
2.4. Evaluation of Apoptosis Induction Induced with Emodin Using Flow Cytometry
2.5. Changes in the Gene and Protein Expression of ER Stress and Apoptosis Markers Induced Using Cotreatment with Emodin and STF-083010
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cells and Cell Culture
4.3. Measurement of Cell Metabolic Activity
4.4. Detection of Morphological Properties (HCS Assay)
4.5. qPCR Analysis
4.6. XBP1 Splicing
4.7. Western Blot Analysis
4.8. Cell Apoptosis Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gajęcki, M.T.; Gajęcka, M.; Zielonka, Ł. The presence of mycotoxins in feed and their influence on animal health. Toxins 2020, 12, 663. [Google Scholar] [CrossRef]
- Wells, J.M.; Cole, R.J.; Kirksey, J.W. Emodin, a toxic metabolite of Aspergillus wentii isolated from weevil-damaged chestnuts. Appl. Microbiol. 1975, 30, 26–28. [Google Scholar] [CrossRef]
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging mycotoxins: Beyond traditionally determined food contaminants. J. Agric. Food Chem. 2017, 65, 7052–7070. [Google Scholar] [CrossRef]
- Sulyok, M.; Krska, R.; Schuhmacher, R. Application of an LC–MS/MS based multi-mycotoxin method for the semi-quantitative determination of mycotoxins occurring in different types of food infected by moulds. Food Chem. 2010, 119, 408–416. [Google Scholar] [CrossRef]
- Khoshal, A.K.; Novak, B.; Martin, P.G.P.; Jenkins, T.; Neves, M.; Schatzmayr, G.; Oswald, I.P.; Pinton, P. Co-occurrence of DON and emerging mycotoxins in worldwide finished pig feed and their combined toxicity in intestinal cells. Toxins 2019, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Ghilardelli, F.; Atzori, A.S.; Zara, S.; Novak, B.; Faas, J.; Fancello, F. Co-occurrence of regulated and emerging mycotoxins in corn silage: Relationships with fermentation quality and bacterial communities. Toxins 2021, 13, 232. [Google Scholar] [CrossRef] [PubMed]
- Na, M.K.; Park, J.Y.; An, R.B.; Lee, S.M.; Kim, Y.H.; Lee, J.P.; Seong, R.S.; Lee, K.S.; Bae, K.H. Quality evaluation of Polygoni Multiflori Radix. Korean J. Pharmacogn. 2000, 31, 335–339. [Google Scholar]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Huyiligeqi; Ni, J. Emodin: A review of its pharmacology, toxicity and pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Xue, J.; Ding, W.; Liu, Y. Anti-diabetic effects of emodin involved in the activation of PPARγ on high-fat diet-fed and low dose of streptozotocin-induced diabetic mice. Fitoterapia 2010, 81, 173–177. [Google Scholar] [CrossRef]
- Ma, J.; Zheng, L.; He, Y.S.; Li, H.J. Hepatotoxic assessment of Polygoni Multiflori Radix extract and toxicokinetic study of stilbene glucoside and anthraquinones in rats. J. Ethnopharmacol. 2015, 162, 61–68. [Google Scholar] [CrossRef]
- Lin, L.; Lin, H.; Zhang, M.; Ni, B.; Yin, X.; Qu, C.; Ni, J. A novel method to analyze hepatotoxic components in Polygonum multiflorum using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. J. Hazard. Mater. 2015, 299, 249–259. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Y.; Qu, Y.; Cheng, M.; Xiao, H. Metabolomic profiling of emodin-induced cytotoxicity in human liver cells and mechanistic study. Toxicol. Res. 2015, 4, 948–955. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Qiu, L.Z.; Yue, L.X.; Ni, Y.H.; Zhou, W.; Huang, C.S.; Deng, H.F.; Wang, N.N.; Liu, H.; Liu, X.; Zhou, Y.Q.; et al. Emodin-induced oxidative inhibition of mitochondrial function assists BiP/IRE1α/CHOP signaling-mediated ER-related apoptosis. Oxid. Med. Cell. Longev. 2021, 2021, 8865813. [Google Scholar] [CrossRef]
- Ming, J.; Ruan, S.; Wang, M.; Ye, D.; Fan, N.; Meng, Q.; Tian, B.; Huang, T. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 2015, 6, 40692–40703. [Google Scholar] [CrossRef]
- Weiskirchen, R. Letter to the Editor: LO2, a misidentified cell line: Some data should be interpreted with caution. Hepatology 2023, 77, E66. [Google Scholar] [CrossRef]
- Chen, C.; Gao, J.; Wang, T.S.; Guo, C.; Yan, Y.J.; Mao, C.Y.; Gu, L.W.; Yang, Y.; Li, Z.F.; Liu, A. NMR-based metabolomic techniques identify the toxicity of emodin in HepG2 cells. Sci. Rep. 2018, 8, 9379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.S.; Wang, X.W.; Sun, Q.F.; Ye, Z.J.; Liu, J.W.; Zhou, D.H.; Tang, Y. Anticancer effects of emodin on HepG2 cell: Evidence from bioinformatic analysis. BioMed Res. Int. 2019, 2019, 3065818. [Google Scholar] [CrossRef]
- Doonan, F.; Cotter, T.G. Morphological assessment of apoptosis. Methods 2008, 44, 200–204. [Google Scholar] [CrossRef]
- Donato, M.T.; Tolosa, L.; Gómez-Lechón, M.J. Culture and functional characterization of human hepatoma HepG2 cells. Methods Mol. Biol. 2015, 1250, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Gorji-Bahri, G.; Moradtabrizi, N.; Hashemi, A. Uncovering the stability status of the reputed reference genes in breast and hepatic cancer cell lines. PLoS ONE 2021, 16, 0259669. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Kim, R.; Erni, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis 2006, 11, 5–13. [Google Scholar] [CrossRef]
- Kwon, K.; Goo, T.W.; Kwon, O.Y. Development of rapid detection method for unfolded protein response in the mammalian cells. J. Exp. Biomed. Sci. 2005, 11, 249–252. [Google Scholar]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef]
- Brady, H.J.M.; Gil-Gómez, G. Molecules in focus Bax. The pro-apoptotic Bcl-2 family member, Bax. Int. J. Biochem. Cell Biol. 1998, 30, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Kaptan, E.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mTORC1. Oncotarget 2017, 8, 68191–68207. [Google Scholar] [CrossRef]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef]
- Barez, S.R.; Atar, A.M.; Aghaei, M. Mechanism of inositol-requiring enzyme 1-alpha inhibition in endoplasmic reticulum stress and apoptosis in ovarian cancer cells. J. Cell Commun. Signal. 2020, 14, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Yang, L.; Yang, Y.; Yang, J.; Niu, Z.; Zhang, X.; Song, Q.; Lei, Y.; Wu, H.; Guo, J. Activation of Wnt/β-catenin pathway causes insulin resistance and increases lipogenesis in HepG2 cells via regulation of endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2020, 526, 764–771. [Google Scholar] [CrossRef]
- Wang, D.; Hou, C.; Cao, Y.; Cheng, Q.; Zhang, L.; Li, H.; Feng, L.; Shen, Y. XBP1 activation enhances MANF expression via binding to endoplasmic reticulum stress response elements within MANF promoter region in hepatitis B. Int. J. Biochem. Cell Biol. 2018, 99, 140–146. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphological Properties | Descriptor | VCON | Emodin 25 μM | Emodin 50 μM | Emodin 100 μM | Emodin 200 μM |
|---|---|---|---|---|---|---|
| Basic intensity and morphology | Cell area | 1.00 ± 0.06 b | 1.02 ± 0.05 b | 1.02 ± 0.07 b | 1.35 ± 0.50 ab | 1.86 ± 0.94 a |
| Cell roundness | 1.00 ± 0.01 | 1.00 ± 0.01 | 1.01 ± 0.01 | 1.01 ± 0.03 | 1.00 ± 0.04 | |
| Nucleus area | 1.00 ± 0.03 a | 0.99 ± 0.03 ab | 0.95 ± 0.03 abc | 0.94 ± 0.08 bc | 0.92 ± 0.05 c | |
| Nucleus roundness | 1.00 ± 0.01 a | 0.98 ± 0.01 ab | 0.96 ± 0.02 bc | 0.94 ± 0.04 cd | 0.92 ± 0.05 d | |
| Nucleus intensity | 1.00 ± 0.04 a | 0.91 ± 0.04 b | 0.87 ± 0.05 b | 0.86 ± 0.09 bc | 0.80 ± 0.06 c | |
| Cytoplasm area | 1.00 ± 0.07 b | 1.03 ± 0.07 b | 1.05 ± 0.09 b | 1.49 ± 0.68 ab | 2.18 ± 1.27 a | |
| Cytoplasm intensity | 1.00 ± 0.06 ab | 1.05 ± 0.04 a | 1.01 ± 0.06 ab | 0.89 ± 0.08 b | 0.68 ± 0.19 c | |
| STAR | Compactness, 60% | 1.00 ± 0.03 | 1.02 ± 0.03 | 1.06 ± 0.03 | 1.03 ± 0.13 | 1.05 ± 0.08 |
| Radial | 1.00 ± 0.03 b | 1.00 ± 0.02 b | 0.98 ± 0.03 b | 1.06 ± 0.09 ab | 1.12 ± 0.14 a | |
| Juxtamembrane | 1.00 ± 0.01 c | 1.01 ± 0.01 bc | 1.01 ± 0.02 bc | 1.03 ± 0.03 ab | 1.05 ± 0.04 a | |
| Cytosolic | 1.00 ± 0.00 a | 1.00 ± 0.00 a | 0.99 ± 0.01 a | 0.98 ± 0.01 b | 0.98 ± 0.01 b | |
| Perinuclear | 1.00 ± 0.01 a | 0.99 ± 0.01 ab | 0.97 ± 0.02 bc | 0.96 ± 0.02 cd | 0.93 ± 0.03 d | |
| Outer nuclear | 1.00 ± 0.02 c | 1.00 ± 0.02 c | 1.02 ± 0.03 bc | 1.07 ± 0.06 a | 1.05 ± 0.05 ab | |
| Inner nuclear | 1.00 ± 0.03 b | 1.02 ± 0.02 b | 1.05 ± 0.03 b | 1.13 ± 0.09 a | 1.12 ± 0.08 a | |
| SER | Spot | 1.00 ± 0.03 b | 0.99 ± 0.02 b | 1.00 ± 0.03 b | 1.06 ± 0.04 b | 1.23 ± 0.15 a |
| Hole | 1.00 ± 0.03 b | 0.99 ± 0.03 b | 1.01 ± 0.03 b | 1.06 ± 0.05 b | 1.24 ± 0.17 a | |
| Edge | 1.00 ± 0.02 a | 0.98 ± 0.02 ab | 0.96 ± 0.05 abc | 0.94 ± 0.05 bc | 0.93 ± 0.07 c | |
| Ridge | 1.00 ± 0.02 b | 0.99 ± 0.02 b | 0.99 ± 0.02 b | 1.02 ± 0.03 b | 1.14 ± 0.09 a | |
| Valley | 1.00 ± 0.02 b | 0.99 ± 0.02 b | 1.00 ± 0.03 b | 1.03 ± 0.03 b | 1.14 ± 0.10 a | |
| Saddle | 1.00 ± 0.02 b | 1.00 ± 0.02 b | 0.99 ± 0.02 b | 1.01 ± 0.02 b | 1.10 ± 0.06 a | |
| Bright | 1.00 ± 0.02 b | 0.99 ± 0.02 b | 1.00 ± 0.03 b | 1.04 ± 0.03 b | 1.18 ± 0.12 a | |
| Dark | 1.00 ± 0.02 b | 0.99 ± 0.02 b | 1.00 ± 0.03 b | 1.05 ± 0.04 b | 1.19 ± 0.13 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.B.; Cho, G.H.; Park, Y.E.; Chun, H.S. Emodin, an Emerging Mycotoxin, Induces Endoplasmic Reticulum Stress-Related Hepatotoxicity through IRE1α–XBP1 Axis in HepG2 Cells. Toxins 2023, 15, 455. https://doi.org/10.3390/toxins15070455
Park SB, Cho GH, Park YE, Chun HS. Emodin, an Emerging Mycotoxin, Induces Endoplasmic Reticulum Stress-Related Hepatotoxicity through IRE1α–XBP1 Axis in HepG2 Cells. Toxins. 2023; 15(7):455. https://doi.org/10.3390/toxins15070455
Chicago/Turabian StylePark, Su Been, Gun Hee Cho, Young Eun Park, and Hyang Sook Chun. 2023. "Emodin, an Emerging Mycotoxin, Induces Endoplasmic Reticulum Stress-Related Hepatotoxicity through IRE1α–XBP1 Axis in HepG2 Cells" Toxins 15, no. 7: 455. https://doi.org/10.3390/toxins15070455
APA StylePark, S. B., Cho, G. H., Park, Y. E., & Chun, H. S. (2023). Emodin, an Emerging Mycotoxin, Induces Endoplasmic Reticulum Stress-Related Hepatotoxicity through IRE1α–XBP1 Axis in HepG2 Cells. Toxins, 15(7), 455. https://doi.org/10.3390/toxins15070455