Modulation of Airway Expression of the Host Bactericidal Enzyme, sPLA2-IIA, by Bacterial Toxins


Abstract
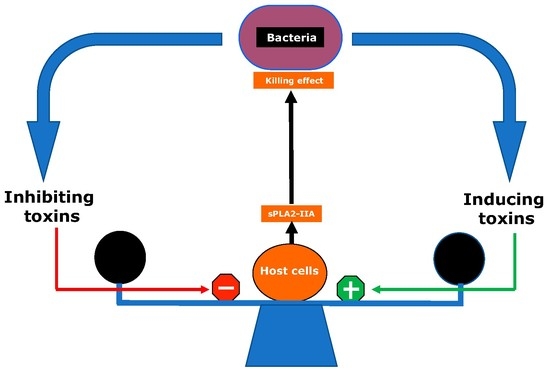
1. Role of sPLA2-IIA in Infectious and Inflammatory Diseases
1.1. General Biological Functions of sPLA2-IIA
1.2. PAMPs, Toxins and Innate Immune Response to Bacterial Infections
1.3. The sPLA2-IIA, an Endogenous Antibiotic-Like Protein of the Host
1.4. Role of the sPLA2-IIA in the Gut Microbiota–Lung Axis
1.5. sPLA2-IIA Levels in Biological Fluids of Infectious and Inflammatory Diseases
2. Bacterial Toxins That Upregulate sPLA2-IIA Expression
2.1. LPS Is the Major Bacterial Toxin Inducing sPLA2-IIA Expression by Host Cells
2.2. Role of Pili in the Induction of sPLA2-IIA Expression in the Airways
2.3. The Type 3 Secretion System (T3SS) Toxin, Exotoxin S (ExoS), Plays a Key Role in sPLA2-IIA Expression in the Airways
2.4. Induction of sPLA2-IIA by Porphyromonas Gingivalis and Relevance to Periodontal Disease
2.5. PGN Is the Main Inducer of sPLA2-IIA Expression by Gram-Positive Bacteria
3. Bacterial Toxins That Downregulate sPLA2-IIA Expression
3.1. Inhibition of sPLA2-IIA Expression by Bacillus Anthracis Toxins
3.1.1. Lethal Toxin Downregulates sPLA2-IIA Expression by AMs
3.1.2. Edema Toxin Impairs sPLA2-IIA Expression by AMs
3.2. Inhibition of sPLA2-IIA Expression by a Bordetella Pertussis AC-Hly Toxin
3.3. S. aureus Adenosine Inhibits sPLA2-IIA Expression and Associated Airway Killing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PAMP | pathogen-associated molecular pattern |
| PRRs | pattern recognition receptors |
| PLA2 | phospholipase A2 |
| sPLA2-IIA | type-IIA secreted phospholipase A2 |
| cPLA2 | cytosolic PLA2 |
| G+ bacterium | Gram-positive bacterium |
| G- bacterium | Gram-negative bacterium |
| LTA | lipoteichoic acid |
| PGN | peptidoglycan |
| LPS | lipopolysaccharides |
| dsDNA | double-stranded DNA |
| ARDS | acute respiratory distress syndrome |
| BALF | bronchoalveolar lavage fluid |
| AMs | alveolar macrophages |
| gpAMs | guinea pig alveolar macrophages |
| BECs | bronchial epithelial cells |
| DPPC | dipalmitoyl-phosphatidylcholine |
| BMVECs | brain microvascular endothelial cells |
| AA | arachidonic acid |
| PGD2 | prostaglandin D2 |
| LOS | lipooligosaccharides |
| T3SS | type 3 secretion system |
| ExoS | exotoxin S |
| CF | cystic fibrosis |
| KLF2 | Krüppel-Like Factor 2 |
| GAP | GTPase activating protein |
| ADPRT | ADP ribosyltransferase |
| MDP | muramyl dipeptide |
| MALP-2 | macrophage-activating lipopeptide 2 kDa |
| CpG | cytosine guanosine dinucleotide |
| ET | edema toxin |
| LT | lethal toxin |
| EF | edema factor |
| PA | protective antigen |
| DN-p38 | dominant negative construct of p38 |
| AC-Hly | adenylate cyclasehemolysin |
| C/EBP | CCAAT/enhancer binding protein |
| cAMP | cyclic adenosine monophosphate |
| AdsA | adenosine synthase A |
References
- Dennis, E.A. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 1997, 22, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Tucker, D.E.; Burchett, S.A.; Leslie, C.C. Properties of the Group IV phospholipase A2 family. Prog. Lipid Res. 2006, 45, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Lambeau, G. Emerging roles of secreted phospholipase A(2) enzymes: An update. Biochimie 2013, 95, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Touqui, L.; Arbibe, L. A role for phospholipase A2 in ARDS pathogenesis. Mol. Med. Today 1999, 5, 244–249. [Google Scholar] [CrossRef]
- Touqui, L.; Alaoui-El-Azher, M. Mammalian secreted phospholipases A2 and their pathophysiological significance in inflammatory diseases. Curr. Mol. Med. 2001, 1, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, T.J.; Graham, G.G.; Scott, K.F. Antibacterial actions of secreted phospholipases A2. Review. Biochim. Biophys. Acta 2008, 1781, 1–9. [Google Scholar] [CrossRef]
- Yagami, T.; Ueda, K.; Asakura, K.; Hata, S.; Kuroda, T.; Sakaeda, T.; Takasu, N.; Tanaka, K.; Gemba, T.; Hori, Y. Human group IIA secretory phospholipase A2 induces neuronal cell death via apoptosis. Mol. Pharm. 2002, 61, 114–126. [Google Scholar] [CrossRef]
- Kugiyama, K.; Ota, Y.; Takazoe, K.; Moriyama, Y.; Kawano, H.; Miyao, Y.; Sakamoto, T.; Soejima, H.; Ogawa, H.; Doi, H.; et al. Circulating levels of secretory type II phospholipase A(2) predict coronary events in patients with coronary artery disease. Circulation 1999, 100, 1280–1284. [Google Scholar] [CrossRef]
- Mattsson, N.; Magnussen, C.G.; Ronnemaa, T.; Mallat, Z.; Benessiano, J.; Jula, A.; Taittonen, L.; Kahonen, M.; Juonala, M.; Viikari, J.S.; et al. Metabolic syndrome and carotid intima-media thickness in young adults: Roles of apolipoprotein B, apolipoprotein A-I, C-reactive protein, and secretory phospholipase A2: The cardiovascular risk in young Finns study. Arter. Thromb. Vasc. Biol. 2010, 30, 1861–1866. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Gelb, M.H. Secretory phospholipase A2: A multifaceted family of proatherogenic enzymes. Curr. Cardiol. Rep. 2009, 11, 445–451. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Pare, A.; Rousseau, M.; Naika, G.S.; Levesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Pruzanski, W.; Lambeau, G.; Lazdunski, M.; Cho, W.; Kopilov, J.; Kuksis, A. Hydrolysis of minor glycerophospholipids of plasma lipoproteins by human group IIA, V and X secretory phospholipases A2. Biochim. Biophys. Acta 2007, 1771, 5–19. [Google Scholar] [CrossRef]
- Fourcade, O.; Simon, M.F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar] [CrossRef]
- van Hensbergen, V.P.; Wu, Y.; van Sorge, N.M.; Touqui, L. Type IIA Secreted Phospholipase A2 in Host Defense against Bacterial Infections. Trends Immunol. 2020, 41, 313–326. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold. Spring Harb. Symp. Quant. Biol. 1989, 54 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89, 107087. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.R.; Furniss, R.C.D.; Goddard, P.J.; Clements, A. Modulation of Host Cell Processes by T3SS Effectors. Curr. Top Microbiol. Immunol. 2018, 416, 73–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef]
- Baker, S.F.; Othman, R.; Wilton, D.C. Tryptophan-containing mutant of human (group IIa) secreted phospholipase A2 has a dramatically increased ability to hydrolyze phosphatidylcholine vesicles and cell membranes. Biochemistry 1998, 37, 13203–13211. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Arbibe, L.; Vial, D.; Rosinski-Chupin, I.; Havet, N.; Huerre, M.; Vargaftig, B.B.; Touqui, L. Endotoxin induces expression of type II phospholipase A2 in macrophages during acute lung injury in guinea pigs: Involvement of TNF-alpha in lipopolysaccharide-induced type II phospholipase A2 synthesis. J. Immunol. 1997, 159, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Pernet, E.; Guillemot, L.; Burgel, P.R.; Martin, C.; Lambeau, G.; Sermet-Gaudelus, I.; Sands, D.; Leduc, D.; Morand, P.C.; Jeammet, L.; et al. Pseudomonas aeruginosa eradicates Staphylococcus aureus by manipulating the host immunity. Nat. Commun. 2014, 5, 5105. [Google Scholar] [CrossRef]
- Chaput, C.; Boneca, I.G. Peptidoglycan detection by mammals and flies. Microbes Infect. 2007, 9, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef]
- Raymond, B.; Leduc, D.; Ravaux, L.; Le Goffic, R.; Candela, T.; Raymondjean, M.; Goossens, P.L.; Touqui, L. Edema toxin impairs anthracidal phospholipase A2 expression by alveolar macrophages. PLoS Pathog. 2007, 3, e187. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef] [PubMed]
- Pernet, E.; Brunet, J.; Guillemot, L.; Chignard, M.; Touqui, L.; Wu, Y. Staphylococcus aureus Adenosine Inhibits sPLA2-IIA-Mediated Host Killing in the Airways. J. Immunol. 2015, 194, 5312–5319. [Google Scholar] [CrossRef]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Proft, T.; Baker, E.N. Pili in Gram-negative and Gram-positive bacteria—Structure, assembly and their role in disease. Cell Mol. Life Sci. 2009, 66, 613–635. [Google Scholar] [CrossRef]
- Touqui, L.; Paya, M.; Thouron, F.; Guiyoule, A.; Zarantonelli, M.L.; Leduc, D.; Wu, Y.; Taha, M.K.; Alonso, J.M. Neisseria meningitidis pili induce type-IIA phospholipase A2 expression in alveolar macrophages. FEBS Lett. 2005, 579, 4923–4927. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Ahmad-Nejad, P.; da Costa, C.; Miethke, T.; Kirschning, C.J.; Hacker, H.; Wagner, H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 2001, 276, 31332–31339. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug. Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef]
- Neuhaus, F.C.; Baddiley, J. A continuum of anionic charge: Structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. [Google Scholar] [CrossRef]
- Koduri, R.S.; Gronroos, J.O.; Laine, V.J.; Le Calvez, C.; Lambeau, G.; Nevalainen, T.J.; Gelb, M.H. Bactericidal properties of human and murine groups I, II, V, X, and XII secreted phospholipases A(2). J. Biol. Chem. 2002, 277, 5849–5857. [Google Scholar] [CrossRef]
- Qu, X.D.; Lehrer, R.I. Secretory phospholipase A2 is the principal bactericide for staphylococci and other gram-positive bacteria in human tears. Infect. Immun. 1998, 66, 2791–2797. [Google Scholar] [CrossRef]
- Murakami, M.; Nakatani, Y.; Atsumi, G.; Inoue, K.; Kudo, I. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 1997, 17, 225–283. [Google Scholar] [CrossRef]
- Weinrauch, Y.; Elsbach, P.; Madsen, L.M.; Foreman, A.; Weiss, J. The potent anti-Staphylococcus aureus activity of a sterile rabbit inflammatory fluid is due to a 14-kD phospholipase A2. J. Clin. Investig. 1996, 97, 250–257. [Google Scholar] [CrossRef]
- Miki, Y.; Taketomi, Y.; Kidoguchi, Y.; Yamamoto, K.; Muramatsu, K.; Nishito, Y.; Park, J.; Hosomi, K.; Mizuguchi, K.; Kunisawa, J.; et al. Group IIA secreted phospholipase A2 controls skin carcinogenesis and psoriasis by shaping the gut microbiota. JCI Insight 2022, 7, e152611. [Google Scholar] [CrossRef] [PubMed]
- Dore, E.; Joly-Beauparlant, C.; Morozumi, S.; Mathieu, A.; Levesque, T.; Allaeys, I.; Duchez, A.C.; Cloutier, N.; Leclercq, M.; Bodein, A.; et al. The interaction of secreted phospholipase A2-IIA with the microbiota alters its lipidome and promotes inflammation. JCI Insight 2022, 7, e152638. [Google Scholar] [CrossRef]
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieers, G.; Guery, B.; Delhaes, L. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front. Cell. Infect. Microbiol. 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Khan, N.; Mendonca, L.; Dhariwal, A.; Fontes, G.; Menzies, D.; Xia, J.; Divangahi, M.; King, I.L. Intestinal dysbiosis compromises alveolar macrophage immunity to Mycobacterium tuberculosis. Mucosal Immunol. 2019, 12, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Sequeira, R.P.; Clarke, T.B. The microbiota protects against respiratory infection via GM-CSF signaling. Nat. Commun. 2017, 8, 1512. [Google Scholar] [CrossRef]
- Pruzanski, W.; Vadas, P. Secretory synovial fluid phospholipase A2 and its role in the pathogenesis of inflammation in arthritis. J. Rheumatol. 1988, 15, 1601–1603. [Google Scholar] [PubMed]
- Nakos, G.; Kitsiouli, E.; Hatzidaki, E.; Koulouras, V.; Touqui, L.; Lekka, M.E. Phospholipases A2 and platelet-activating-factor acetylhydrolase in patients with acute respiratory distress syndrome. Crit. Care Med. 2005, 33, 772–779. [Google Scholar] [CrossRef]
- Touqui, L.; Herpin-Richard, N.; Gene, R.M.; Jullian, E.; Aljabi, D.; Hamberger, C.; Vargaftig, B.B.; Dessange, J.F. Excretion of platelet activating factor-acetylhydrolase and phospholipase A2 into nasal fluids after allergenic challenge: Possible role in the regulation of platelet activating factor release. J. Allergy Clin. Immunol. 1994, 94, 109–119. [Google Scholar] [CrossRef]
- Weinrauch, Y.; Abad, C.; Liang, N.S.; Lowry, S.F.; Weiss, J. Mobilization of potent plasma bactericidal activity during systemic bacterial challenge. Role of group IIA phospholipase A2. J. Clin. Investig. 1998, 102, 633–638. [Google Scholar] [CrossRef]
- Gimenez, A.P.; Wu, Y.Z.; Paya, M.; Delclaux, C.; Touqui, L.; Goossens, P.L. High bactericidal efficiency of type iia phospholipase A2 against Bacillus anthracis and inhibition of its secretion by the lethal toxin. J. Immunol. 2004, 173, 521–530. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Kuwata, H.; Yoshihara, K.; Masuda, S.; Shimbara, S.; Oh-ishi, S.; Murakami, M.; Kudo, I. Induction of distinct sets of secretory phospholipase A(2) in rodents during inflammation. Biochim. Biophys. Acta 2003, 1635, 37–47. [Google Scholar] [CrossRef]
- Medjane, S.; Raymond, B.; Wu, Y.; Touqui, L. Impact of CFTR DeltaF508 mutation on prostaglandin E2 production and type IIA phospholipase A2 expression by pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L816–L824. [Google Scholar] [CrossRef] [PubMed]
- Hatzidaki, E.; Nakos, G.; Galiatsou, E.; Lekka, M.E. Impaired phospholipases A₂production by stimulated macrophages from patients with acute respiratory distress syndrome. Biochim. Biophys. Acta 2010, 1802, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Arbibe, L.; Koumanov, K.; Vial, D.; Rougeot, C.; Faure, G.; Havet, N.; Longacre, S.; Vargaftig, B.B.; Bereziat, G.; Voelker, D.R.; et al. Generation of lyso-phospholipids from surfactant in acute lung injury is mediated by type-II phospholipase A2 and inhibited by a direct surfactant protein A-phospholipase A2 protein interaction. J. Clin. Investig. 1998, 102, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Arita, H. Inflammatory factors stimulate expression of group II phospholipase A2 in rat cultured astrocytes. Two distinct pathways of the gene expression. J. Biol. Chem. 1991, 266, 9956–9960. [Google Scholar] [CrossRef]
- Crowl, R.M.; Stoller, T.J.; Conroy, R.R.; Stoner, C.R. Induction of phospholipase A2 gene expression in human hepatoma cells by mediators of the acute phase response. J. Biol. Chem. 1991, 266, 2647–2651. [Google Scholar] [CrossRef]
- Furue, S.; Kuwabara, K.; Mikawa, K.; Nishina, K.; Shiga, M.; Maekawa, N.; Ueno, M.; Chikazawa, Y.; Ono, T.; Hori, Y.; et al. Crucial role of group IIA phospholipase A(2) in oleic acid-induced acute lung injury in rabbits. Am. J. Respir. Crit. Care Med. 1999, 160, 1292–1302. [Google Scholar] [CrossRef]
- Kitsiouli, E.; Antoniou, G.; Gotzou, H.; Karagiannopoulos, M.; Basagiannis, D.; Christoforidis, S.; Nakos, G.; Lekka, M.E. Effect of azithromycin on the LPS-induced production and secretion of phospholipase A2 in lung cells. Biochim. Biophys. Acta 2015, 1852, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qian, P.; Xu, Z.; Zhang, J.; Wang, Y.; Cheng, S.; Cai, W.; Qian, G.; Wang, C.; Decoster, M.A. Regulatory effects of the JAK3/STAT1 pathway on the release of secreted phospholipase A₂-IIA in microvascular endothelial cells of the injured brain. J. Neuroinflamm. 2012, 9, 170. [Google Scholar] [CrossRef]
- Taha, M.K.; Alonso, J.M. Molecular epidemiology of infectious diseases: The example of meningococcal disease. Res. Microbiol. 2008, 159, 62–66. [Google Scholar] [CrossRef]
- Dunn, K.L.; Virji, M.; Moxon, E.R. Investigations into the molecular basis of meningococcal toxicity for human endothelial and epithelial cells: The synergistic effect of LPS and pili. Microb. Pathog. 1995, 18, 81–96. [Google Scholar] [CrossRef]
- O’Grady, E.P.; Mulcahy, H.; O’Callaghan, J.; Adams, C.; O’Gara, F. Pseudomonas aeruginosa infection of airway epithelial cells modulates expression of Kruppel-like factors 2 and 6 via RsmA-mediated regulation of type III exoenzymes S and Y. Infect. Immun. 2006, 74, 5893–5902. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Yang, V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef] [PubMed]
- Dach, K.; Zovko, J.; Hogardt, M.; Koch, I.; van Erp, K.; Heesemann, J.; Hoffmann, R. Bacterial toxins induce sustained mRNA expression of the silencing transcription factor klf2 via inactivation of RhoA and Rhophilin 1. Infect. Immun. 2009, 77, 5583–5592. [Google Scholar] [CrossRef]
- Goehring, U.M.; Schmidt, G.; Pederson, K.J.; Aktories, K.; Barbieri, J.T. The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J. Biol. Chem. 1999, 274, 36369–36372. [Google Scholar] [CrossRef]
- Okuda, J.; Hayashi, N.; Tanabe, S.; Minagawa, S.; Gotoh, N. Degradation of interleukin 8 by the serine protease MucD of Pseudomonas aeruginosa. J. Infect. Chemother. 2011, 17, 782–792. [Google Scholar] [CrossRef]
- Vareechon, C.; Zmina, S.E.; Karmakar, M.; Pearlman, E.; Rietsch, A. Pseudomonas aeruginosa Effector ExoS Inhibits ROS Production in Human Neutrophils. Cell Host Microbe 2017, 21, 611–618. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, A.; Alimova, Y.; Kirakodu, S.; Kozal, A.; Novak, M.J.; Stromberg, A.J.; Orraca, L.; Gonzalez-Martinez, J.; Martinez, M.; Ebersole, J.L.; et al. Activation of Notch-1 in oral epithelial cells by P. gingivalis triggers the expression of the antimicrobial protein PLA(2)-IIA. Mucosal. Immunol. 2018, 11, 1047–1059. [Google Scholar] [CrossRef]
- Potempa, J.; Sroka, A.; Imamura, T.; Travis, J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: Structure, function and assembly of multidomain protein complexes. Curr. Protein Pept. Sci. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R.; Gupta, D. Peptidoglycan recognition in innate immunity. J. Endotoxin Res. 2005, 11, 304–310. [Google Scholar] [CrossRef]
- Glomski, I.J.; Fritz, J.H.; Keppler, S.J.; Balloy, V.; Chignard, M.; Mock, M.; Goossens, P.L. Murine splenocytes produce inflammatory cytokines in a MyD88-dependent response to Bacillus anthracis spores. Cell. Microbiol. 2007, 9, 502–513. [Google Scholar] [CrossRef]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef]
- Lincoln, R.E.; Hodges, D.R.; Klein, F.; Mahlandt, B.G.; Jones, W.I., Jr.; Haines, B.W.; Rhian, M.A.; Walker, J.S. Role of the lymphatics in the pathogenesis of anthrax. J. Infect. Dis. 1965, 115, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, P.C. Anthrax vaccines: Past, present and future. Vaccine 1991, 9, 533–539. [Google Scholar] [CrossRef]
- Raymond, B.; Ravaux, L.; Mémet, S.; Wu, Y.; Sturny-Leclère, A.; Leduc, D.; Denoyelle, C.; Goossens, P.L.; Payá, M.; Raymondjean, M.; et al. Anthrax lethal toxin down-regulates type-IIA secreted phospholipase A(2) expression through MAPK/NF-kappaB inactivation. Biochem. Pharm. 2010, 79, 1149–1155. [Google Scholar] [CrossRef]
- Menschikowski, M.; Hagelgans, A.; Kostka, H.; Eisenhofer, G.; Siegert, G. Involvement of epigenetic mechanisms in the regulation of secreted phospholipase A2 expressions in Jurkat leukemia cells. Neoplasia 2008, 10, 1195–1203. [Google Scholar] [CrossRef]
- Voth, D.E.; Hamm, E.E.; Nguyen, L.G.; Tucker, A.E.; Salles, I.I.; Ortiz-Leduc, W.; Ballard, J.D. Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell. Microbiol. 2005, 7, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Antonio, V.; Brouillet, A.; Janvier, B.; Monne, C.; Bereziat, G.; Andreani, M.; Raymondjean, M. Transcriptional regulation of the rat type IIA phospholipase A2 gene by cAMP and interleukin-1beta in vascular smooth muscle cells: Interplay of the CCAAT/enhancer binding protein (C/EBP), nuclear factor-kappaB and Ets transcription factors. Biochem. J. 2002, 368, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Cleret-Buhot, A.; Mathieu, J.; Tournier, J.N.; Quesnel-Hellmann, A. Both lethal and edema toxins of Bacillus anthracis disrupt the human dendritic cell chemokine network. PLoS ONE 2012, 7, e43266. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Raymond, B.; Goossens, P.L.; Njamkepo, E.; Guiso, N.; Paya, M.; Touqui, L. Type-IIA secreted phospholipase A2 is an endogenous antibiotic-like protein of the host. Biochimie 2010, 92, 583–587. [Google Scholar] [CrossRef]
- Skopova, K.; Tomalova, B.; Kanchev, I.; Rossmann, P.; Svedova, M.; Adkins, I.; Bibova, I.; Tomala, J.; Masin, J.; Guiso, N.; et al. Cyclic AMP-Elevating Capacity of Adenylate Cyclase Toxin-Hemolysin Is Sufficient for Lung Infection but Not for Full Virulence of Bordetella pertussis. Infect. Immun. 2017, 85, e00937-16. [Google Scholar] [CrossRef]
- Foster, T.J. Immune evasion by staphylococci. Nat Rev Microbiol 2005, 3, 948–958. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Lindsay, J.A. Genetic variation in Staphylococcus aureus surface and immune evasion genes is lineage associated: Implications for vaccine design and host-pathogen interactions. BMC Microbiol. 2010, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, J.N.; Gorshkov, B.; Varn, M.N.; Zemskova, M.A.; Zemskov, E.A.; Sridhar, S.; Lucas, R.; Verin, A.D. Protective effect of adenosine receptors against lipopolysaccharide-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L497–L507. [Google Scholar] [CrossRef]
- Takahashi, N.; Tetsuka, T.; Uranishi, H.; Okamoto, T. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur. J. Biochem. 2002, 269, 4559–4565. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| PAMPs | Bacterium | Host PRRs | Effect on sPLA2-IIA Expression | References | |
|---|---|---|---|---|---|
| gpAMs | BECs | ||||
| LPS | G− | TLR4 a | Upregulation b | No effect c | a [21], b [22], c [23] |
| Peptidoglycan | G+; G− | NOD1 a, NOD2 a | Upregulation b | / | a [24], [25], b [26] |
| Lipoteichoic acid | G+ | TLR2 a | No effect b | / | a [27], b [28] |
| Flagellin | G− | TLR5 a | No effect b | No effect c | a [29], b our unpublished data, c [28] |
| Pili | G+; G− | CD46, CD48, CD55, etc a | Upregulation b | No effect c | a [30], b [31], c [23] |
| HSP60 | G+; G− | TLR2, TLR4 a | / | / | a [32] |
| CpG DNA | G+, G− | TLR9 a | / | No effect b | a [33], b [23] |
| HSL | G− | / | / | No effect a | a [23] |
| ExoS | G− | / | / | Upregulation a | a [23] |
| Adenosine | G+ | Adenosine receptor a | downregulation b | / | a [34], b [28] |
| AC-Hly | G− | CD11b/CD18 integrin a | downregulation b | / | a [35], b [28] |
| Edema toxin | G+ | CMG2 a, TEM8 a | downregulation b | / | a [36], [37], b [26] |
| Lethal toxin | G+ | CMG2 a, TEM8 a | downregulation b | / | a [36], [37], b [26] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Pernet, E.; Touqui, L. Modulation of Airway Expression of the Host Bactericidal Enzyme, sPLA2-IIA, by Bacterial Toxins. Toxins 2023, 15, 440. https://doi.org/10.3390/toxins15070440
Wu Y, Pernet E, Touqui L. Modulation of Airway Expression of the Host Bactericidal Enzyme, sPLA2-IIA, by Bacterial Toxins. Toxins. 2023; 15(7):440. https://doi.org/10.3390/toxins15070440
Chicago/Turabian StyleWu, Yongzheng, Erwan Pernet, and Lhousseine Touqui. 2023. "Modulation of Airway Expression of the Host Bactericidal Enzyme, sPLA2-IIA, by Bacterial Toxins" Toxins 15, no. 7: 440. https://doi.org/10.3390/toxins15070440
APA StyleWu, Y., Pernet, E., & Touqui, L. (2023). Modulation of Airway Expression of the Host Bactericidal Enzyme, sPLA2-IIA, by Bacterial Toxins. Toxins, 15(7), 440. https://doi.org/10.3390/toxins15070440