Integration of Transcriptomics and Microbiomics Reveals the Responses of Bellamya aeruginosa to Toxic Cyanobacteria


Abstract
1. Introduction
2. Results
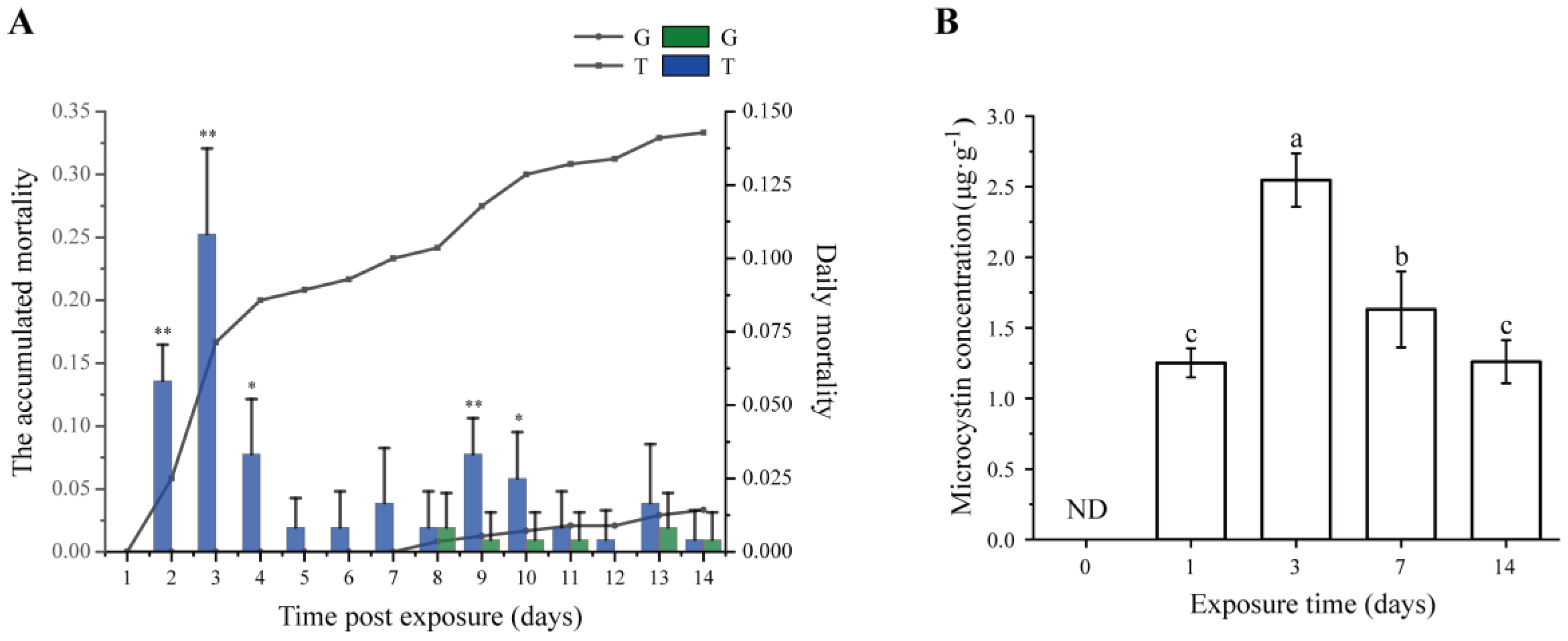
2.1. Snail Mortality
2.2. Dynamics of MC Concentration
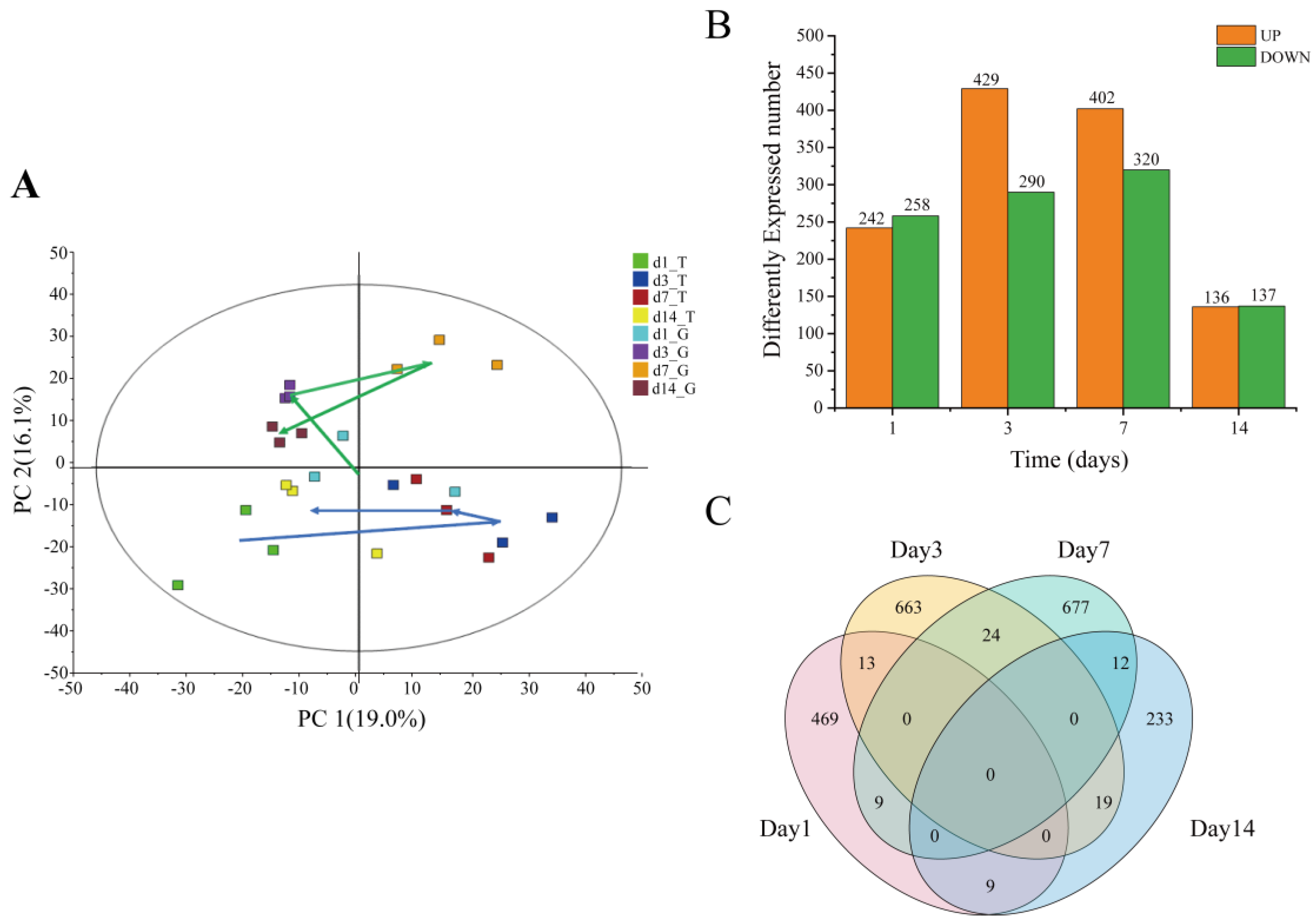
2.3. Transcriptomic Analysis of Hepatopancreas
2.3.1. GO Functional Annotation of DEGs
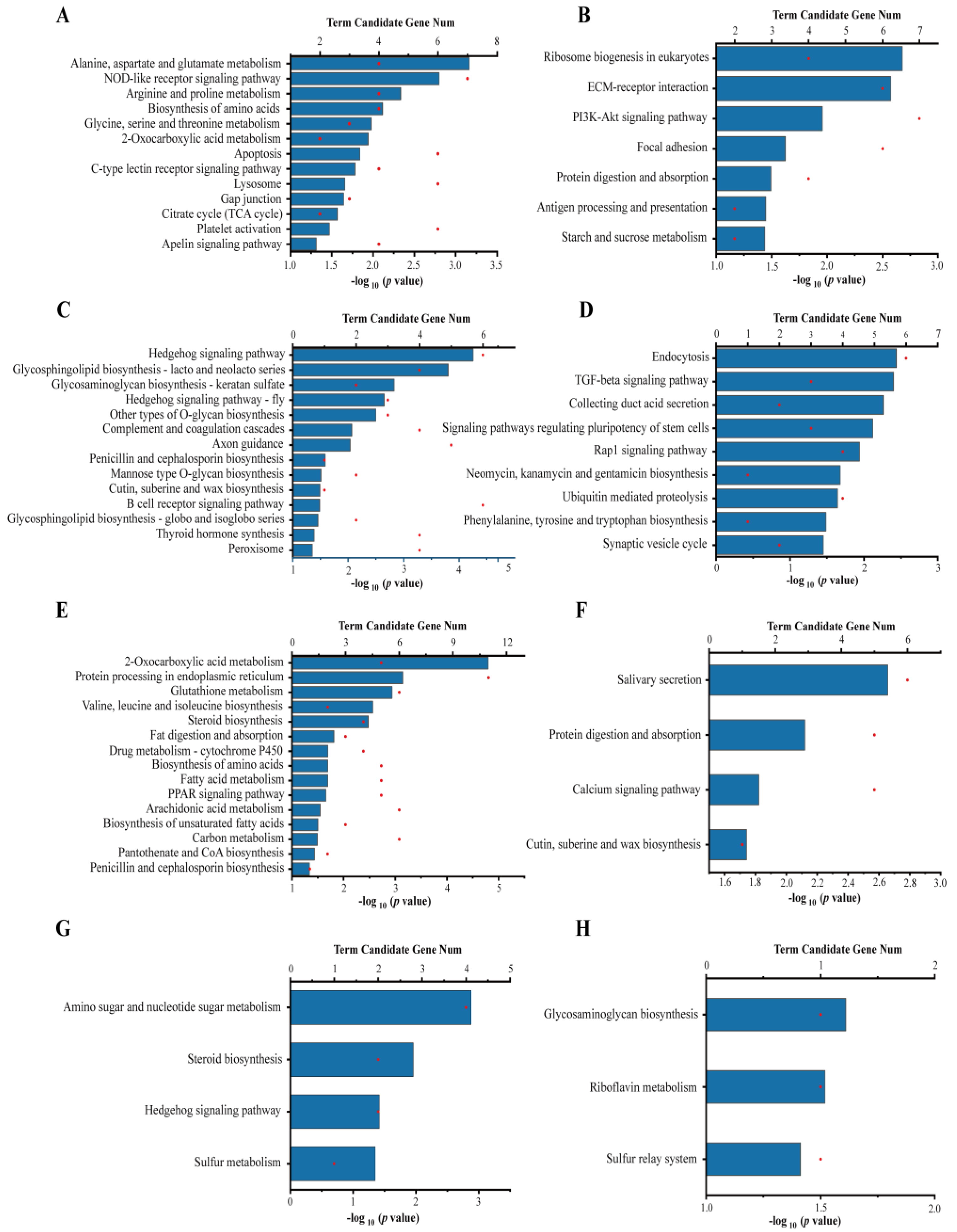
2.3.2. KEGG Pathway Enrichment Analysis of DEGs
2.3.3. Identification of Host Health-Related DEGs
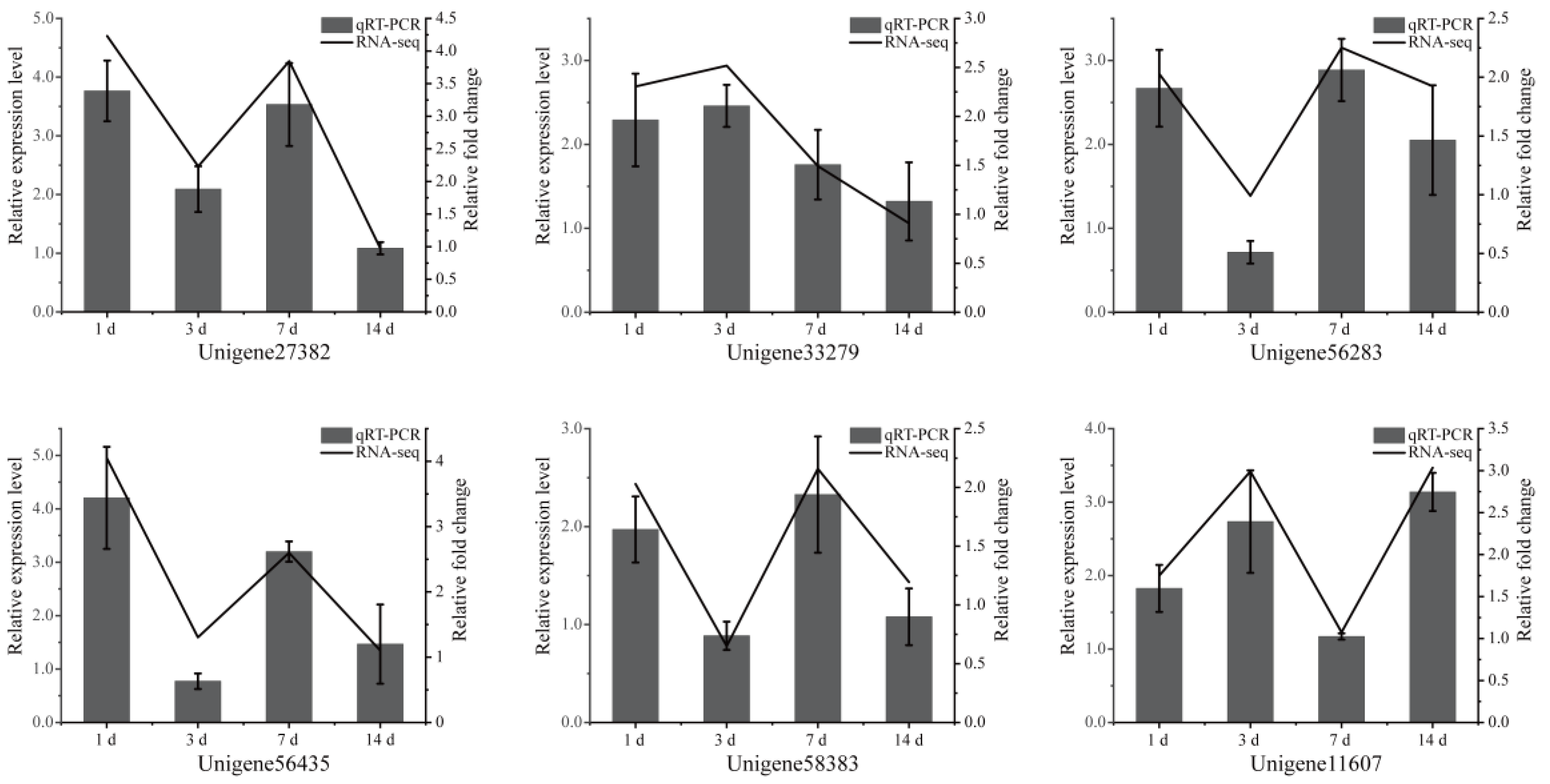
2.3.4. Validation of Gene Expression in the Transcriptome
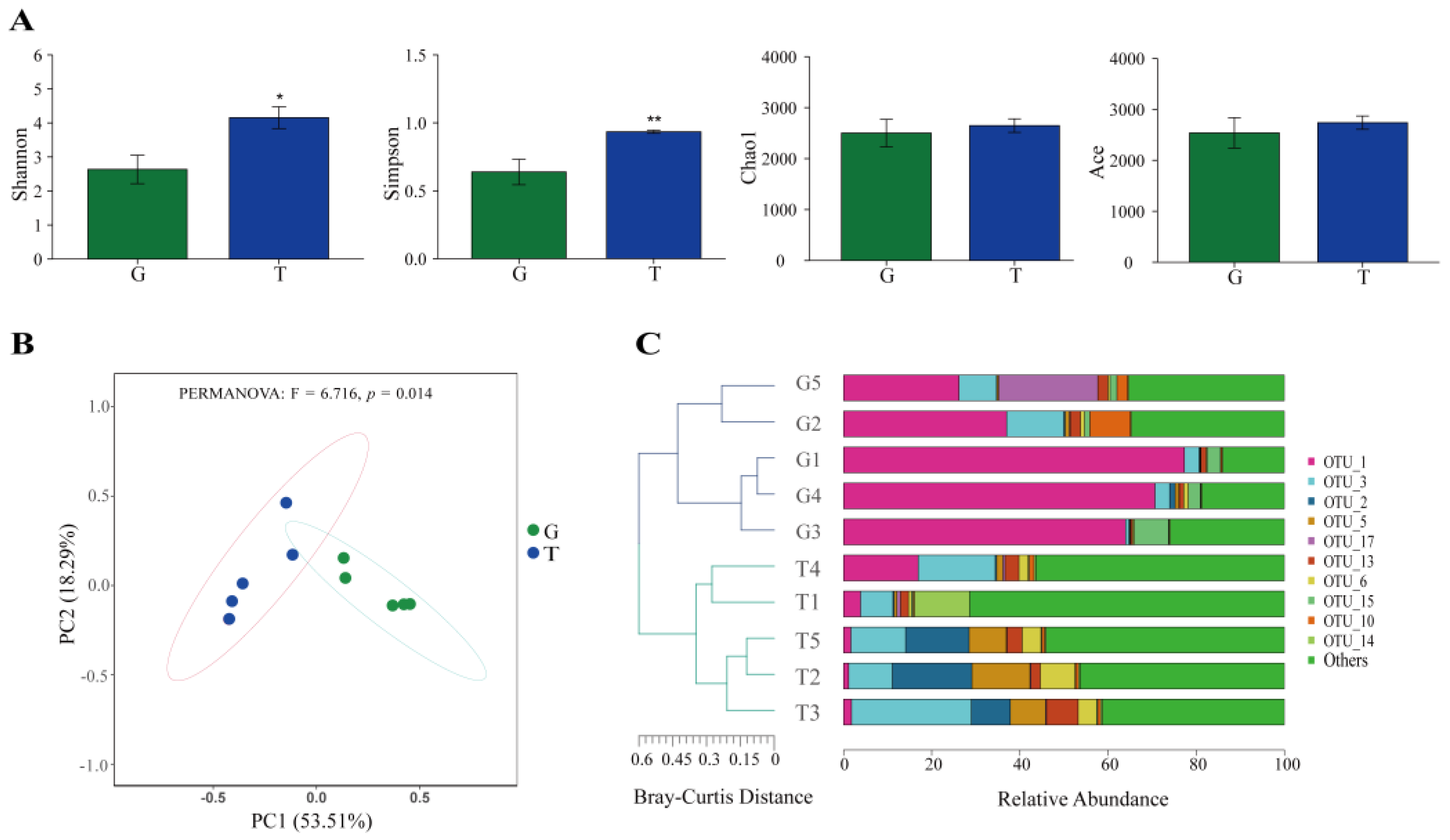
2.4. Intestinal Microbiota Analysis
2.4.1. Effects of Toxic Cyanobacteria on the Intestinal Microbiota of B. aeruginosa
2.4.2. Co-Occurrence Networks
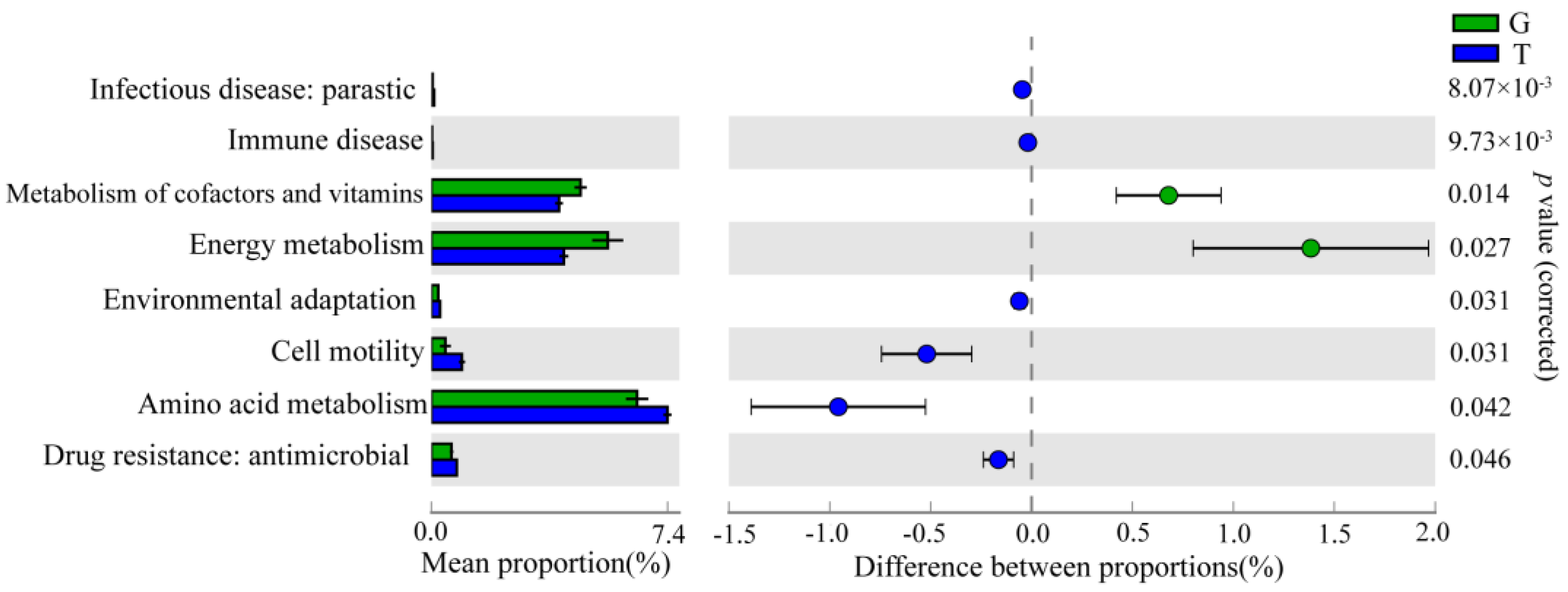
2.4.3. Functional Prediction Analysis of Intestinal Microbiota
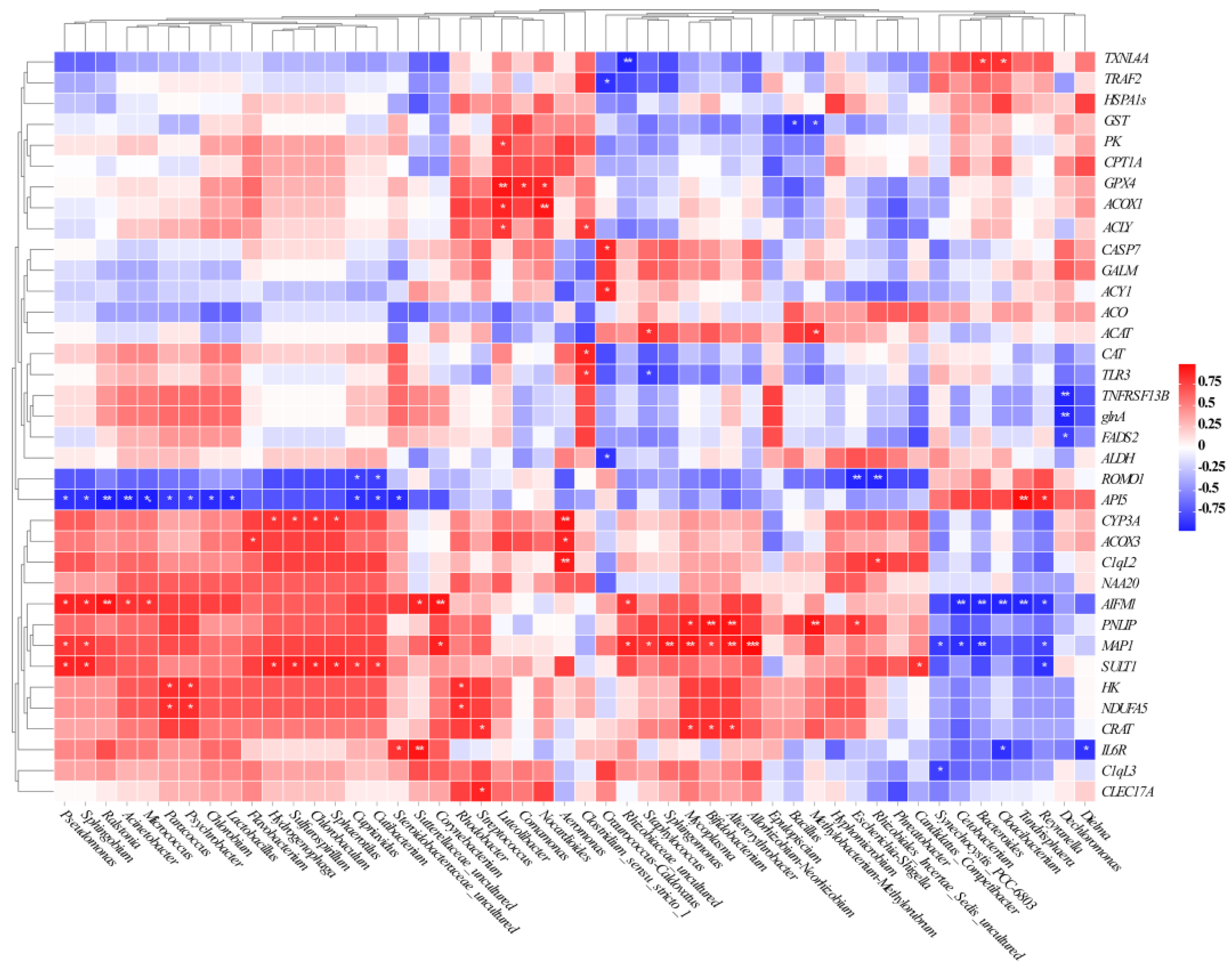
2.5. Association between the Intestinal Microbiota and the DEGs Related to Host Health
3. Discussion
3.1. Hepatopancreatic Transcriptomic Response to Toxic Cyanobacteria
3.1.1. Stress Responses and Xenobiotics Biodegradation
3.1.2. Immune Responses
3.1.3. Energy Metabolism
3.2. Intestinal Microbiota in Response to Toxic Cyanobacterial Stress
3.3. Relationship between Gut Microbial and Host Health-Related DEGs
4. Conclusions
5. Materials and Methods
5.1. Test Snail and Algae Cultivation
5.2. Experimental Design and Sample Collection
5.3. MC Determination
5.4. Transcriptomic Analysis of Hepatopancreas
5.5. qRT-PCR Validation
5.6. DNA Extraction, Amplification, and Illumina Hiseq
5.7. Correlation Analysis of Gut Microbiota and DEGs
5.8. Statistical and Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mowe, M.A.; Mitrovic, S.M.; Lim, R.P.; Furey, A.; Yeo, D.C. Tropical cyanobacterial blooms: A review of prevalence, problem taxa, toxins and influencing environmental factors. J. Limnol. 2015, 74, 205–224. [Google Scholar] [CrossRef]
- Bláha, L.; Babica, P.; Maršálek, B. Toxins produced in cyanobacterial water blooms–toxicity and risks. Interdiscip. Toxicol. 2009, 2, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xie, P.; Liu, Y.; Qiu, T. Transfer, distribution and bioaccumulation of microcystins in the aquatic food web in Lake Taihu, China, with potential risks to human health. Sci. Total Environ. 2009, 407, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Bownik, A. Harmful algae: Effects of cyanobacterial cyclic peptides on aquatic invertebrates—A short review. Toxicon 2016, 124, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, C.; Pflugmacher, S. Ecotoxicological effects of selected cyanobacterial secondary metabolites a short review. Toxicol. Appl. Pharmacol. 2005, 203, 201–218. [Google Scholar] [CrossRef]
- Drobac, D.; Tokodi, N.; Lujić, J.; Marinović, Z.; Subakov-Simić, G.; Dulić, T.; Važić, T.; Nybom, S.; Meriluoto, J.; Codd, G.A. Cyanobacteria and cyanotoxins in fishponds and their effects on fish tissue. Harmful Algae 2016, 55, 66–76. [Google Scholar] [CrossRef]
- Neves, R.A.; Nascimento, S.M.; Santos, L.N. Harmful algal blooms and shellfish in the marine environment: An overview of the main molluscan responses, toxin dynamics, and risks for human health. Environ. Sci. Pollut. Res. 2021, 28, 55846–55868. [Google Scholar] [CrossRef]
- Agasild, H.; Panksep, K.; Tõnno, I.; Blank, K.; Kõiv, T.; Freiberg, R.; Laugaste, R.; Jones, R.I.; Nõges, P.; Nõges, T. Role of potentially toxic cyanobacteria in crustacean zooplankton diet in a eutrophic lake. Harmful Algae 2019, 89, 101688. [Google Scholar] [CrossRef]
- Lance, E.; Desprat, J.; Holbech, B.F.; Gerard, C.; Bormans, M.; Lawton, L.A.; Edwards, C.; Wiegand, C. Accumulation and detoxication responses of the gastropod Lymnaea stagnalis to single and combined exposures to natural (cyanobacteria) and anthropogenic (the herbicide RoundUp® Flash) stressors. Aquat. Toxicol. 2016, 177, 116–124. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, K.; Zhang, C.; Liang, J.; Hu, Z. Biochemical and ultrastructural changes in the hepatopancreas of Bellamya aeruginosa (Gastropoda) fed with toxic cyanobacteria. Sci. World J. 2011, 11, 2091–2105. [Google Scholar] [CrossRef]
- AL-Sultan, E.Y.A. Isolation, Purification and Identification of Blue-green Alga Hapalosiphon aureus and Evaluation of its Histopathological Effects on Fresh Water Snail Lymnaea auricularia. J. Appl. Sci. 2017, 17, 61–71. [Google Scholar] [CrossRef]
- Qiao, F.; Lei, K.; Li, Z.; Wei, Z.; Liu, Q.; Yang, L.; He, J.; An, L.; Qi, H.; Cui, S. Transcriptomic responses of the freshwater snail (Parafossarulus striatulus) following dietary exposure to cyanobacteria. Sci. Total Environ. 2018, 624, 153–161. [Google Scholar] [CrossRef]
- Yang, W.; Ye, Y.; Lu, K.; Zheng, Z.; Zhu, J. NMR-based metabolomic responses of freshwater gastropod Bellamya aeruginosa to MC-producing and non MC-producing Microcystis aeruginosa. J. Oceanol. Limnol. 2022, 40, 260–272. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, Z.; Wang, Z.; Zhang, J.; Xie, Z.; Wang, Z. Oxidative stress responses and toxin accumulation in the freshwater snail Radix swinhoei (Gastropoda, Pulmonata) exposed to microcystin-LR. Environ. Sci. Pollut. Res. 2016, 23, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hu, Y.; He, J.; Chen, J.; Giesy, J.P.; Xie, P. Responses of the Proteome and Metabolome in Livers of Zebrafish Exposed Chronically to Environmentally Relevant Concentrations of Microcystin-LR. Environ. Sci. Technol. 2017, 51, 596–607. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, H.; Du, X.; Shi, Z.; Liu, X.; Wang, R.; Zhang, S.; Tian, Z.; Shi, L.; Guo, H.; et al. Advances in the toxicology research of microcystins based on Omics approaches. Environ. Int. 2021, 154, 106661. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, J.; Liu, S.; Wang, C.; Jin, Y.; Lai, H.; Tu, W. Aberrant hepatic lipid metabolism associated with gut microbiota dysbiosis triggers hepatotoxicity of novel PFOS alternatives in adult zebrafish. Environ. Int. 2022, 166, 107351. [Google Scholar] [CrossRef]
- Fu, Z.; Han, F.; Huang, K.; Zhang, J.; Qin, J.G.; Chen, L.; Li, E. Impact of imidacloprid exposure on the biochemical responses, transcriptome, gut microbiota and growth performance of the Pacific white shrimp Litopenaeus vannamei. J. Hazard. Mater. 2022, 424, 127513. [Google Scholar] [CrossRef]
- Weingarten, E.A.; Atkinson, C.L.; Jackson, C.R. The gut microbiome of freshwater Unionidae mussels is determined by host species and is selectively retained from filtered seston. PLoS ONE 2019, 14, e0224796. [Google Scholar] [CrossRef]
- North, E.; Minton, R.L. Diversity and Predicted Function of Gut Microbes from Two Species of Viviparid Snails. Freshw. Mollusk Biol. Conserv. 2021, 24, 104–113. [Google Scholar] [CrossRef]
- Wijanarka, W.; Kusdiyantini, E.; Parman, S. Screening cellulolytic bacteria from the digestive tract snail (Achatina fulica) and test the ability of cellulase activity. Biosaintifika J. Biol. Biol. Educ. 2016, 8, 385–391. [Google Scholar] [CrossRef]
- Takacs-Vesbach, C.; King, K.; Van Horn, D.; Larkin, K.; Neiman, M. Distinct bacterial microbiomes in sexual and asexual Potamopyrgus antipodarum, a New Zealand freshwater snail. PloS ONE 2016, 11, e0161050. [Google Scholar] [CrossRef]
- Dushku, E.; Ioannou, A.; Staikou, A.; Yiangou, M. Probiotic properties and immunomodulatory activity of gastrointestinal tract commensal bacterial strains isolated from the edible farmed snail Cornu aspersum maxima. Fish Shellfish Immunol. 2019, 92, 792–801. [Google Scholar] [CrossRef]
- Macke, E.; Callens, M.; De Meester, L.; Decaestecker, E. Host-genotype dependent gut microbiota drives zooplankton tolerance to toxic cyanobacteria. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- SCHULTHEIß, R.; Wilke, T.; JØRGENSEN, A.; Albrecht, C. The birth of an endemic species flock: Demographic history of the Bellamya group (Gastropoda, Viviparidae) in Lake Malawi. Biol. J. Linn. Soc. 2011, 102, 130–143. [Google Scholar] [CrossRef]
- Van Bocxlaer, B.; Hunt, G. Morphological stasis in an ongoing gastropod radiation from Lake Malawi. Proc. Natl. Acad. Sci. USA 2013, 110, 13892–13897. [Google Scholar] [CrossRef]
- Stelbrink, B.; Richter, R.; Köhler, F.; Riedel, F.; Strong, E.E.; Van Bocxlaer, B.; Albrecht, C.; Hauffe, T.; Page, T.J.; Aldridge, D.C. Global diversification dynamics since the Jurassic: Low dispersal and habitat-dependent evolution explain hotspots of diversity and shell disparity in river snails (Viviparidae). Syst. Biol. 2020, 69, 944–961. [Google Scholar] [CrossRef]
- Lance, E.; Brient, L.; Bormans, M.; Gérard, C. Interactions between cyanobacteria and Gastropods: I. Ingestion of toxic Planktothrix agardhii by Lymnaea stagnalis and the kinetics of microcystin bioaccumulation and detoxification. Aquat. Toxicol. 2006, 79, 140–148. [Google Scholar] [CrossRef]
- Qu, M.; Qu, Y.; Ren, W.; Wang, Y.; Wu, Q. The mechanism of controlling Microcystis bloom by Bellamya aeruginosa. J. Fudan Univ. Nat. Sci. 2010, 49, 301–308. [Google Scholar] [CrossRef]
- Lyu, T.; Zhu, J.; Yang, X.; Yang, W.; Zheng, Z. Responses of Gut Microbial Community Composition and Function of the Freshwater Gastropod Bellamya aeruginosa to Cyanobacterial Bloom. Front. Microbiol. 2022, 13, 906278. [Google Scholar] [CrossRef]
- Lance, E.; Josso, C.; Dietrich, D.; Ernst, B.; Paty, C.; Senger, F.; Bormans, M.; Gerard, C. Histopathology and microcystin distribution in Lymnaea stagnalis (Gastropoda) following toxic cyanobacterial or dissolved microcystin-LR exposure. Aquat. Toxicol. 2010, 98, 211–220. [Google Scholar] [CrossRef]
- Ferrao-Filho Ada, S.; Kozlowsky-Suzuki, B. Cyanotoxins: Bioaccumulation and effects on aquatic animals. Mar. Drugs 2011, 9, 2729–2772. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, K.; Zheng, Z.; Pan, J.; Zhu, J. Accumulation and degradation of microcystins and their effects on activities of three kinds of enzymes in liver of Bellamya aeruginosa. J. Agro-Environ. Sci. 2009, 28, 54–59. [Google Scholar] [CrossRef]
- Baganz, D.; Staaks, G.; Steinberg, C. Impact of the cyanobacteria toxin, microcystin-lr on behaviour of zebrafish, Danio rerio. Water Res. 1998, 32, 948–952. [Google Scholar] [CrossRef]
- Qu, X.; Hu, M.; Shang, Y.; Pan, L.; Jia, P.; Fu, C.; Liu, Q.; Wang, Y. Liver transcriptome and miRNA analysis of silver carp (Hypophthalmichthys molitrix) intraperitoneally injected with microcystin-LR. Front. Physiol. 2018, 9, 381. [Google Scholar] [CrossRef]
- Qiao, Q.; Le Manach, S.; Huet, H.; Duvernois-Berthet, E.; Chaouch, S.; Duval, C.; Sotton, B.; Ponger, L.; Marie, A.; Mathéron, L. An integrated omic analysis of hepatic alteration in medaka fish chronically exposed to cyanotoxins with possible mechanisms of reproductive toxicity. Environ. Pollut. 2016, 219, 119–131. [Google Scholar] [CrossRef]
- Wei, L.; Hoole, D.; Sun, B. Identification of apoptosis-related genes and transcription variations in response to microcystin-LR in zebrafish liver. Toxicol. Ind. Health 2014, 30, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Brzuzan, P.; Florczyk, M.; Łakomiak, A.; Woźny, M. Illumina sequencing reveals aberrant expression of microRNAs and their variants in whitefish (Coregonus lavaretus) liver after exposure to microcystin-LR. PLoS ONE 2016, 11, e0158899. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Fu, J.; He, L.; Wang, H.; Ruan, J.; Li, F.; Wu, H. Microcystin-LR-induced autophagy regulates oxidative stress, inflammation, and apoptosis in grass carp ovary cells in vitro. Toxicol. In Vitro 2023, 87, 105520. [Google Scholar] [CrossRef]
- Wang, P.; Peng, X.; Wei, Z.-F.; Wei, F.-Y.; Wang, W.; Ma, W.-D.; Yao, L.-P.; Fu, Y.-J.; Zu, Y.-G. Geraniin exerts cytoprotective effect against cellular oxidative stress by upregulation of Nrf2-mediated antioxidant enzyme expression via PI3K/AKT and ERK1/2 pathway. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 1751–1761. [Google Scholar] [CrossRef]
- Pflugmacher, S.; Wiegand, C.; Oberemm, A.; Beattie, K.A.; Krause, E.; Codd, G.A.; Steinberg, C.E.W. Identification of an enzymatically formed glutathione conjugate of the cyanobacterial hepatotoxin microcystin-LR: The first step of detoxication. Biochim. Biophys. Acta BBA Gen. Subj. 1998, 1425, 527–533. [Google Scholar] [CrossRef]
- Hoffmann, U.; Kroemer, H.K. The ABC transporters MDR1 and MRP2: Multiple functions in disposition of xenobiotics and drug resistance. Drug Metab. Rev. 2004, 36, 669–701. [Google Scholar] [CrossRef]
- Hégaret, H.; Da Silva, P.M.; Wikfors, G.H.; Haberkorn, H.; Shumway, S.E.; Soudant, P. In vitro interactions between several species of harmful algae and haemocytes of bivalve molluscs. Cell Biol. Toxicol. 2011, 27, 249–266. [Google Scholar] [CrossRef]
- Diez-Quijada, L.; Benítez-González, M.d.M.; Puerto, M.; Jos, A.; Cameán, A.M. Immunotoxic Effects Induced by Microcystins and Cylindrospermopsin: A Review. Toxins 2021, 13, 711. [Google Scholar] [CrossRef]
- Gorbushin, A.M. Immune response of a caenogastropod host: A case study of Littorina littorea and its digenean parasites. Dev. Comp. Immunol. 2019, 101, 103465. [Google Scholar] [CrossRef]
- Seppälä, O.; Walser, J.-C.; Cereghetti, T.; Seppälä, K.; Salo, T.; Adema, C.M. Transcriptome profiling of Lymnaea stagnalis (Gastropoda) for ecoimmunological research. BMC Genomics 2021, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.A.; Elkon, K.B. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr. Dir. Autoimmun. 2006, 9, 120–142. [Google Scholar] [CrossRef]
- Court, N.; Vasseur, V.; Vacher, R.; Frémond, C.; Shebzukhov, Y.; Yeremeev, V.V.; Maillet, I.; Nedospasov, S.A.; Gordon, S.; Fallon, P.G.; et al. Partial Redundancy of the Pattern Recognition Receptors, Scavenger Receptors, and C-Type Lectins for the Long-Term Control of Mycobacterium tuberculosis Infection. J. Immunol. 2010, 184, 7057–7070. [Google Scholar] [CrossRef]
- Conus, S.; Simon, H.-U. Cathepsins and their involvement in immune responses. Swiss Med. Wkly. 2010, 140, w13042. [Google Scholar] [CrossRef]
- King, K.L.; Cidlowski, J.A. Cell cycle and apoptosis: Common pathways to life and death. J. Cell. Biochem. 1995, 58, 175–180. [Google Scholar] [CrossRef]
- Ansaldo, M.; Nahabedian, D.E.; Holmes-Brown, E.; Agote, M.; Ansay, C.V.; Guerrero, N.R.V.; Wider, E.A. Potential use of glycogen level as biomarker of chemical stress in Biomphalaria glabrata. Toxicology 2006, 224, 119–127. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.X.; Wu, B.; Yin, J.; Yu, Y.; Yang, L. Comprehensive insights into microcystin-LR effects on hepatic lipid metabolism using cross-omics technologies. J. Hazard. Mater. 2016, 315, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Lu, Z.; Zeng, S.; Dan, X.; Mo, Z.; Zhang, J.; Li, Y. Integration of intestinal microbiota and transcriptomic and metabolomic responses reveals the toxic responses of Litopenaeus vannamei to microcystin-LR. Ecotoxicol. Environ. Saf. 2021, 228, 113030. [Google Scholar] [CrossRef]
- Li, G.; Li, J.; Kohl, K.D.; Yin, B.; Wei, W.; Wan, X.; Zhu, B.; Zhang, Z. Dietary shifts influenced by livestock grazing shape the gut microbiota composition and co-occurrence networks in a local rodent species. J. Anim. Ecol. 2019, 88, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Massey, I.Y.; Yang, F. A mini review on microcystins and bacterial degradation. Toxins 2020, 12, 268. [Google Scholar] [CrossRef]
- Riaz Rajoka, M.S.; Wu, Y.; Mehwish, H.M.; Bansal, M.; Zhao, L. Lactobacillus exopolysaccharides: New perspectives on engineering strategies, physiochemical functions, and immunomodulatory effects on host health. Trends Food Sci. Technol. 2020, 103, 36–48. [Google Scholar] [CrossRef]
- Lowe-Power, T.M.; Khokhani, D.; Allen, C. How Ralstonia solanacearum exploits and thrives in the flowing plant xylem environment. Trends Microbiol. 2018, 26, 929–942. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Ng, T.H.; Wu, J.-H.; Chen, J.-W.; Wang, H.-C. Microbiome dynamics in a shrimp grow-out pond with possible outbreak of acute hepatopancreatic necrosis disease. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Santolini, M.; Barabási, A.-L. Predicting perturbation patterns from the topology of biological networks. Proc. Natl. Acad. Sci. USA 2018, 115, E6375–E6383. [Google Scholar] [CrossRef]
- Winter, S.E.; Bäumler, A.J. Why related bacterial species bloom simultaneously in the gut: Principles underlying the ‘Like will to like’concept. Cell. Microbiol. 2014, 16, 179–184. [Google Scholar] [CrossRef]
- Silveira, M.A.D.; Bilodeau, S.; Greten, T.F.; Wang, X.W.; Trinchieri, G. The gut-liver axis: Host microbiota interactions shape hepatocarcinogenesis. Trends Cancer 2022, 8, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut–Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef]
- Huang, J.M.; Hong, H.A.; Van Tong, H.; Hoang, T.H.; Brisson, A.; Cutting, S.M. Mucosal delivery of antigens using adsorption to bacterial spores. Vaccine 2010, 28, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, X.; Wu, Z.; Xiong, S.; Zhou, Z.; Wang, X.; Xu, J.; Lu, F.; Yu, X. Immunogenicity of self-adjuvanticity oral vaccine candidate based on use of Bacillus subtilis spore displaying Schistosoma japonicum 26 KDa GST protein. Parasitol. Res. 2009, 105, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, G.; Arciola, C.R.; Rindi, S.; Di Poto, A.; Missineo, A.; Montanaro, L.; Speziale, P. Toll-like receptors (TLRs) in innate immune defense against Staphylococcus aureus. Int. J. Artif. Organs 2011, 34, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; Dockrell, D.H.; Pattery, T.; Lee, D.G.; Cornelis, P.; Hellewell, P.G.; Whyte, M.K. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J. Immunol. 2005, 174, 3643–3649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, Y.; Ansari, A.R.; Akhtar, M.; Chen, Y.; Cheng, R.; Cui, L.; Nafady, A.A.; Elokil, A.A.; Abdel-Kafy, E.S.M. Caecal microbiota could effectively increase chicken growth performance by regulating fat metabolism. Microb. Biotechnol. 2022, 15, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Basta, T.; Keck, A.; Klein, J.; Stolz, A. Detection and characterization of conjugative degradative plasmids in xenobiotic-degrading Sphingomonas strains. J. Bacteriol. 2004, 186, 3862–3872. [Google Scholar] [CrossRef]
- Yoshida, N.; Yamashita, T.; Osone, T.; Hosooka, T.; Shinohara, M.; Kitahama, S.; Sasaki, K.; Sasaki, D.; Yoneshiro, T.; Suzuki, T.; et al. Bacteroides spp. promotes branched-chain amino acid catabolism in brown fat and inhibits obesity. iScience 2021, 24, 103342. [Google Scholar] [CrossRef]
- Ahmad, M.T.; Shariff, M.; Md Yusoff, F.; Goh, Y.M.; Banerjee, S. Applications of microalga Chlorella vulgaris in aquaculture. Rev. Aquacult. 2020, 12, 328–346. [Google Scholar] [CrossRef]
- Hu, L.; Shan, K.; Lin, L.; Shen, W.; Huang, L.; Gan, N.; Song, L. Multi-Year Assessment of Toxic Genotypes and Microcystin Concentration in Northern Lake Taihu, China. Toxins 2016, 8, 23. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID a | NR Annotation | KO Name | log2 (Fold Change) b | |||
|---|---|---|---|---|---|---|
| 1 d | 3 d | 7 d | 14 d | |||
| Oxidative response/xenobiotics biodegradation | ||||||
| Unigene33914 | thioredoxin-like protein 4B [Pomacea canaliculata] | TXNL4A | 1.18 * | −0.54 | 1.51 ** | −0.03 |
| CL3662.Contig2 | cytochrome P450 3A43-like [Pomacea canaliculata] | CYP3A | 0.79 | 0.85 | 1.54 * | 1.20 ** |
| CL3025.Contig1 | catalase-like isoform X1 [Pomacea canaliculata] | CAT | 0.22 | 1.60 ** | 1.06 * | 0.33 |
| Unigene25071 | sulfotransferase 1A3-like isoform X2 [Pomacea canaliculata] | SULT1 | −0.38 | 1.18 | 1.24 ** | 1.78 ** |
| CL9050.Contig1 | pi-class glutathione S-transferase [Cipangopaludina cathayensis] | GST | 0.81 | −0.46 | 1.14 * | 1.55 ** |
| CL515.Contig1 | glutathione peroxidase-like [Pomacea canaliculata] | GPX4 | 0.92 | −0.14 | 1.13 * | 1.25 ** |
| CL5413.Contig1 | heat shock protein 70 [Mytilus coruscus] | HSPA1s | −0.36 | 1.06 ** | 1.19 ** | 0.11 |
| CL8943.Contig2 | reactive oxygen species modulator 1-like isoform X1 [Anneissia japonica] | ROMO1 | 1.31 ** | -0.44 | −1.09 ** | −0.24 |
| Immune responses | ||||||
| CL7726.Contig2 | interleukin-6 receptor subunit beta-like isoform X2 [Pomacea canaliculata] | IL6R | 1.13 ** | 0.31 | 2.31 ** | 1.03 |
| Unigene58383 | TNF receptor-associated factor 2-like isoform X1 [Pomacea canaliculata] | TRAF2 | 1.02 * | -0.62 | 1.11 * | 0.26 |
| Unigene43614 | Toll-like receptor 8 [Pomacea canaliculata] | TLR3 | 1.40 * | -0.33 | 1.34 * | 0.38 |
| CL10774.Contig2 | C-type lectin domain family 4 member M-like [Pomacea canaliculata] | CLEC17A | −0.79 | 1.16 * | 1.24 * | 0.84 |
| CL10094.Contig1 | tumor necrosis factor receptor superfamily member 13B-like [Pomacea canaliculata] | TNFRSF13B | 2.01 ** | 1.93 ** | 1.66 | 0.24 |
| CL1261.Contig2 | caspase-3-like [Pomacea canaliculata] | CASP7 | −0.32 | 0.71 | 1.54 ** | 1.15 * |
| CL4333.Contig1 | apoptosis-inducing factor 1, mitochondrial-like isoform X3 [Pomacea canaliculata] | AIFM1 | 1.71 * | 0.38 | 0.13 | 1.52 * |
| Unigene46579 | apoptosis inhibitor 5-like [Pomacea canaliculata] | API5 | 1.62 * | 1.03 * | -0.15 | -0.81 |
| Unigene44570 | complement C1q-like protein 3 [Pomacea canaliculata] | C1qL3 | −0.53 | 1.16 ** | 1.06 | 1.61 ** |
| Unigene56138 | complement C1q-like protein 2 [Pomacea canaliculata] | C1qL2 | −1.30 * | 0.29 | 4.01 ** | 1.19 |
| Energy metabolism | ||||||
| Unigene9677 | aconitate hydratase, mitochondrial-like [Aplysia californica] | ACO | −0.84 | 1.20 * | −1.68 ** | - |
| Unigene53307 | ATP-citrate synthase-like isoform X2 [Pomacea canaliculata] | ACLY | 1.13 * | -0.07 | 1.06 * | 0.75 |
| CL11869.Contig1 | hexokinase type 2-like [Pomacea canaliculata] | HK | 2.39 ** | - | 1.02 | 2.10 ** |
| CL4947.Contig1 | aldehyde dehydrogenase family 3 member B1-like isoform X5 [Pomacea canaliculata] | ALDH | −0.73 | 1.88 * | 1.09 * | 0.49 |
| Unigene44545 | pyruvate kinase PKM-like isoform X4 [Pomacea canaliculata] | PK | 2.01 ** | 0.16 | 1.09 ** | 0.53 |
| Unigene61388 | aldose 1-epimerase-like [Pomacea canaliculata] | GALM | 1.56 | −1.78 ** | 0.24 | 1.84 * |
| CL10453.Contig2 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5-like [Pomacea canaliculata] | NDUFA5 | −1.52 | 7.35 ** | 1.30 ** | 1.63 |
| Unigene63153 | pancreatic triacylglycerol lipase-like [Pomacea canaliculata] | PNLIP | 0.34 | 0.61 | 1.69 * | 1.26 * |
| Unigene34763 | peroxisomal acyl-coenzyme A oxidase 1-like [Pomacea canaliculata] | ACOX1 | 1.57 * | 0.01 | 1.56 ** | 0.47 |
| Unigene56283 | peroxisomal acyl-coenzyme A oxidase 3-like [Pomacea canaliculata] | ACOX3 | 1.02 * | -0.01 | 1.17 * | 0.94 |
| CL10117.Contig4 | fatty acid desaturase 2-like isoform X1 [Pomacea canaliculata] | FADS2 | 2.00 ** | 1.13 | 3.11 ** | 0.52 |
| CL8795.Contig2 | acetyl-CoA acetyltransferase, mitochondrial-like [Pomacea canaliculata] | ACAT | −1.21 * | 1.91 ** | 1.43 | 0.07 |
| Unigene43422 | carnitine O-palmitoyltransferase 1, liver isoform-like isoform X2 [Pomacea canaliculata] | CPT1A | 2.14 ** | 1.24 * | 0.63 | 0.15 |
| CL6049.Contig1 | glutamine synthetase-like [Pomacea canaliculata] | glnA | 1.34 * | 1.65 * | 1.06 | 0.46 |
| CL2579.Contig1 | aminoacylase-1-like [Pomacea canaliculata] | ACY1 | 1.39 * | 0.10 | 1.08 * | 0.23 |
| CL5226.Contig1 | carnitine O-acetyltransferase-like isoform X1 [Pomacea canaliculata] | CRAT | 0.83 | 1.51 * | 0.33 | 1.66 * |
| CL3812.Contig4 | N-alpha-acetyltransferase 20-like [Pomacea canaliculata] | NAA20 | 0.34 | 2.07 * | 1.63 * | 0.85 |
| Unigene26308 | methionine aminopeptidase 1-like isoform X1 [Pecten maximus] | MAP1 | −0.85 | 1.33 * | −0.26 | 1.44 * |
| Index | G | T |
|---|---|---|
| node | 192 | 289 |
| edge | 617 | 1467 |
| avgK | 6.427 | 10.152 |
| avgCC | 0.72 | 0.733 |
| positive/negative (%) | 90.11/9.89 | 85.55/14.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhu, J.; Hu, C.; Yang, W.; Zheng, Z. Integration of Transcriptomics and Microbiomics Reveals the Responses of Bellamya aeruginosa to Toxic Cyanobacteria. Toxins 2023, 15, 119. https://doi.org/10.3390/toxins15020119
Yang X, Zhu J, Hu C, Yang W, Zheng Z. Integration of Transcriptomics and Microbiomics Reveals the Responses of Bellamya aeruginosa to Toxic Cyanobacteria. Toxins. 2023; 15(2):119. https://doi.org/10.3390/toxins15020119
Chicago/Turabian StyleYang, Xianming, Jinyong Zhu, Chaoyang Hu, Wen Yang, and Zhongming Zheng. 2023. "Integration of Transcriptomics and Microbiomics Reveals the Responses of Bellamya aeruginosa to Toxic Cyanobacteria" Toxins 15, no. 2: 119. https://doi.org/10.3390/toxins15020119
APA StyleYang, X., Zhu, J., Hu, C., Yang, W., & Zheng, Z. (2023). Integration of Transcriptomics and Microbiomics Reveals the Responses of Bellamya aeruginosa to Toxic Cyanobacteria. Toxins, 15(2), 119. https://doi.org/10.3390/toxins15020119