1. Introduction
Molds of the genera
Aspergillus,
Penicillium,
Fusarium, and
Alternaria are known to produce a broad spectrum of structurally different mycotoxins that can contaminate various types of crops and food commodities and induce a variety of toxicological effects [
1]. The contamination of food by these fungal toxic metabolites occurs in both developed and developing countries, with the latter generally showing higher contamination levels [
2]. However, due to growing globalization and climate change enabling more favorable conditions for the growth of these fungi, an increased occurrence of such toxic metabolites in food is also expected in the near future in developed countries [
3].
Among the various mycotoxins known to date, those produced by the genus
Alternaria are still not regulated due to the shortage of toxicological and occurrence data which prevents a proper risk assessment from being conducted. In fact, unlike other mycotoxins, for which their maximum levels in food have been set by the Commission Regulation (EU) 2023/915 [
4], only indicative levels are currently available for certain
Alternaria mycotoxins in food. Of note, the exceedance of these levels only implies the need to investigate the factors leading to the presence of the mycotoxins or the effects of food processing (Commission Recommendation (EU) 2022/553) [
5].
Alternaria species produce mixtures of secondary metabolites that can be classified into five groups, namely dibenzo-α-pyrones, perylene quinones, tetramic acid derivatives,
Alternaria alternata f. sp.
lycopersici toxins, and miscellaneous structures [
1]. In this context, the mycotoxin alternariol (AOH), which belongs to the dibenzo-α-pyrones group, is one of the most studied
Alternaria mycotoxins. It can be found in a wide variety of foods, including grains, tomato, fruits (and their respective products), sunflower seeds/oil, and fermented beverages like beer and wine [
1,
6]. It is important to highlight that data on food occurrences consistently identify AOH as one of the most commonly detected
Alternaria mycotoxins in food, with food often experiencing simultaneous contamination by multiple
Alternaria mycotoxins [
1]. Of note, a previous study suggested that the actual occurrence may even exceed the present estimate [
7]. With respect to its toxicological properties, AOH has been extensively reported to exert estrogenic effects [
8,
9] as well as to induce different types of DNA damage, including single-strand breaks (SSB), double-strand breaks (DSB), and oxidative DNA damage through mechanisms that involve the generation of reactive oxygen species and the poisoning of DNA topoisomerases (TOPOs), especially the IIα isoform [
10]. TOPOs are a family of enzymes that regulate the DNA topological state by breaking one (TOPO I) or both (TOPO II) strands of the DNA which are subsequently rejoined. Thus, the mycotoxin AOH is able to stabilize the DNA–topoisomerase complex that is formed during DNA replication or transcription, thus inhibiting the re-ligation of the DNA which results in the persistence of single- (TOPO I poisoning) and double- (TOPO II poisoning) strand breaks [
10,
11]. AOH was also reported to act as a mutagenic compound, being able to induce hypoxanthine-guanine phosphoribosyltransferase (HPRT) mutations in V79 cells and thymidine kinase (TK) gene mutations in mouse lymphoma cells [
12].
TOPO activity has also been reported to be affected by some process contaminants, including acrylamide (AA) [
13]. AA is a small hydrophilic compound primarily formed in carbohydrate-rich foods, such as baked products, through the Maillard reaction during thermal processing at high temperatures (equal to or higher than 120 °C) [
14,
15]. In vivo studies have reported AA to be quickly absorbed from the gastro-intestinal (GI) tract, to be distributed throughout the whole body and extensively metabolized by conjugation with glutathione or epoxidation to glycidamide (GA) [
16,
17]. The genotoxicity and mutagenic potential of AA and its reactive metabolite GA have been studied extensively both in vitro and in vivo. In particular, an in vitro study found AA to induce the formation of DNA adducts (mostly N7-guanine adducts), which can lead to depurination or mispairings and, therefore, an increased mutation rate during DNA replication [
18]. AA itself has been shown to induce chromosomal aberrations, micronuclei, and other mitotic disturbances in mammalian cells [
19]. Based on the scientific data available, the International Agency for Research on Cancer (IARC) has classified AA as a “probable human carcinogen” (group 2A) [
16]. Similar to AOH, there are currently no established maximum levels in food for AA. However, the Commission Regulation (EU) 2017/2158 provides benchmark levels for AA, which are not safety levels and only serve as performance indicators. Exceeding these benchmark levels triggers food business operators to review mitigation measures [
20]. With respect to the ability of AA to target TOPOs, in vitro studies have found AA to act as a catalytic inhibitor of TOPO II in V79 Chinese hamster cells [
13], thus acting differently from the mycotoxin AOH. In fact, unlike TOPO poisons, catalytic inhibitors bind to the enzyme and interfere with its ability to cleave and re-ligate DNA strands without stabilizing the protein–DNA cleavage complexes.
Apart from the previously mentioned food commodities, AOH has been found to occur in several thermally processed cereal-based products, such as wheat and rye bread, breakfast cereals, cookies, and biscuits, which are known to often contain AA [
21,
22,
23]. Despite this, cereal-based products are not among the most contaminated commodities by AOH. Indeed, the concentrations and the frequency of occurrence of AOH in these products are generally much lower than those found in other foods [
1]. Nevertheless, the co-exposure of humans to both contaminants remains highly probable. In the context of a varied diet, the consumption of various foods during a meal could, in fact, expose consumers to both contaminants, even if they are present in different food items.
Building upon this foundation and considering their well-known capacity to induce genotoxic and mutagenic effects [
1,
6,
16], the primary objective of the present study was to explore their potential combined genotoxic and mutagenic effects in vitro, which could potentially arise following the consumption of foods contaminated with these widely present compounds. To achieve this and to assess the impact of metabolic activation on the toxicity of the individual compounds and combinations, the In-Cell Western assay of γ-H2AX (genotoxicity) and the Ames microplate format test (mutagenicity) were performed in the presence and absence of the rat S9 fraction.
3. Discussion
The emerging
Alternaria mycotoxin AOH has gained increasing interest from the scientific community over the past few decades due to its high occurrence in food and broad spectrum of adverse effects reported in vitro and in vivo [
6]. However, since data currently available are still not sufficient to conduct a comprehensive risk assessment, no regulations are currently in place. AOH was reported to occur in several cereal-based foods, including bakery products [
21,
22]. These products are known to be often contaminated by the process contaminant AA, whose production occurs at a high temperature of cooking because of the Maillard reaction [
24]. Despite the co-occurrence of AOH and AA in food and their well-known individual genotoxic and mutagenic properties [
1,
6,
16], there is currently a lack of knowledge regarding the potential toxicological outcomes on human health associated with their combination. Therefore, the aim of the present study was to assess, for the first time, the possible onset of combinatory genotoxic and mutagenic effects resulting from the co-exposure to these compounds. In addition, the possible occurrence of combinatory cytotoxic effects was also investigated.
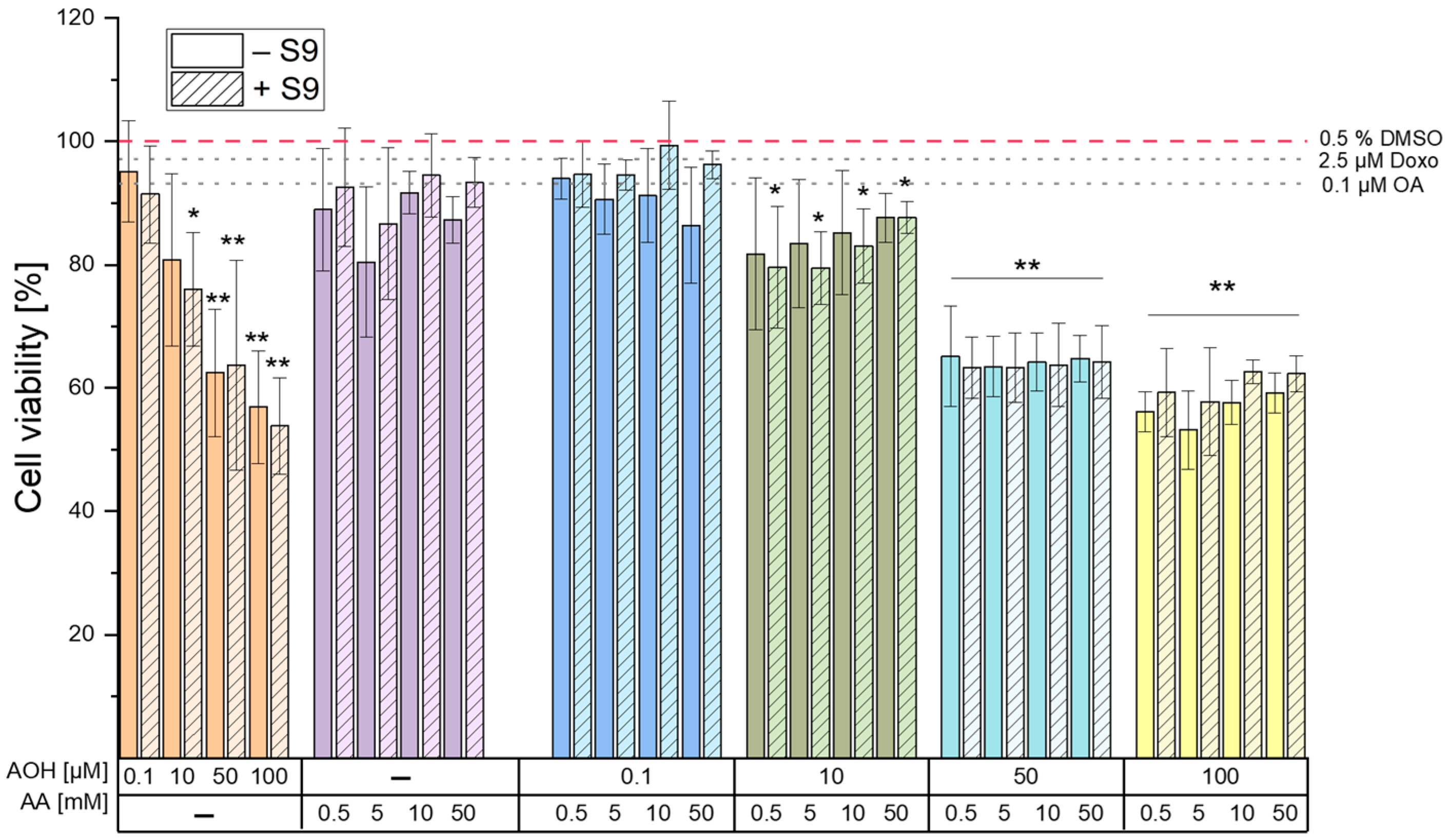
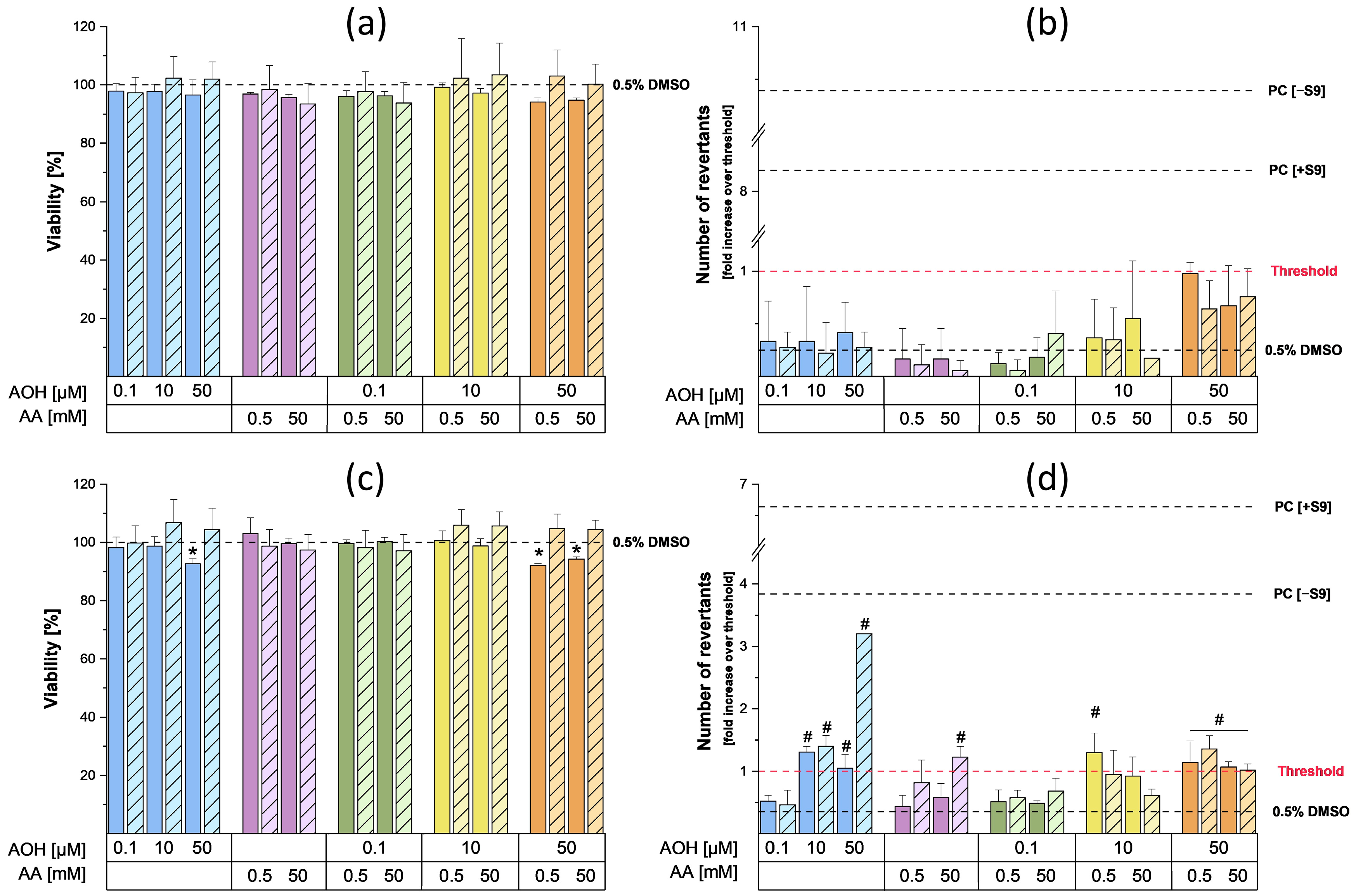
The results of the cell viability assay deriving from a 4 h exposure of HepG2 cells to different concentrations of AOH and AA (as single compounds or in combination) revealed the inability of AA to induce cytotoxic effects up to the highest concentration tested (50 mM). On the contrary, AOH was found to decrease the viability of HepG2 cells in a concentration-dependent manner (
Figure 1) starting from a concentration of 10 µM (in the presence of metabolic activation). The exposure of cells to 100 µM AOH resulted in viabilities of 53.9 ± 7.8% and 56.9 ± 9.2% in the presence and absence of metabolic activation, respectively. Of note, a decrease in the viability of HepG2 cells to 53 ± 13% was also reported by Hessel-Pras et al. [
25] after the exposure of the cells to 100 µM AOH for the same incubation time. Furthermore, Vejdovszky et al. [
26] showed the ability of AOH to induce a dose-dependent cytotoxic effect in the HepG2 cell line (EC50: 51.4 µM), with no further significant decrease in cell viability at concentrations higher than 50 µM. These results are in accordance with those obtained in the present study, in which the exposure of cells to 50 µM and 100 µM AOH resulted in a similar cell viability (62 ± 10.3% and 56.9 ± 9.2%, respectively). As shown in
Figure 1, the treatment of cells with binary combinations of AOH and AA resulted in decreased cell viability without any observable combinatory effects. In fact, the changes in cell viability almost completely overlapped with those induced by the respective concentrations of AOH tested individually, thus suggesting AOH-driven cytotoxic effects.
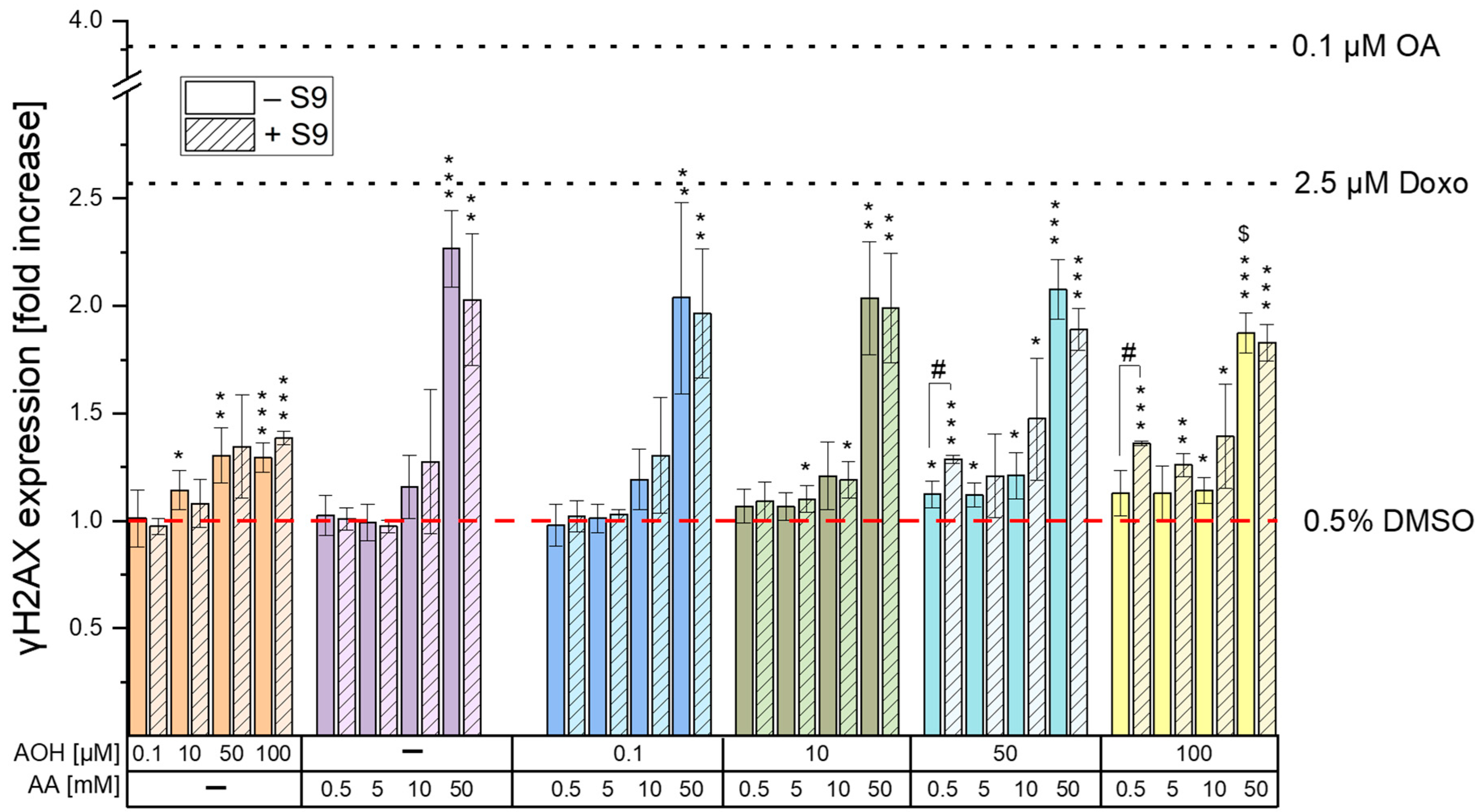
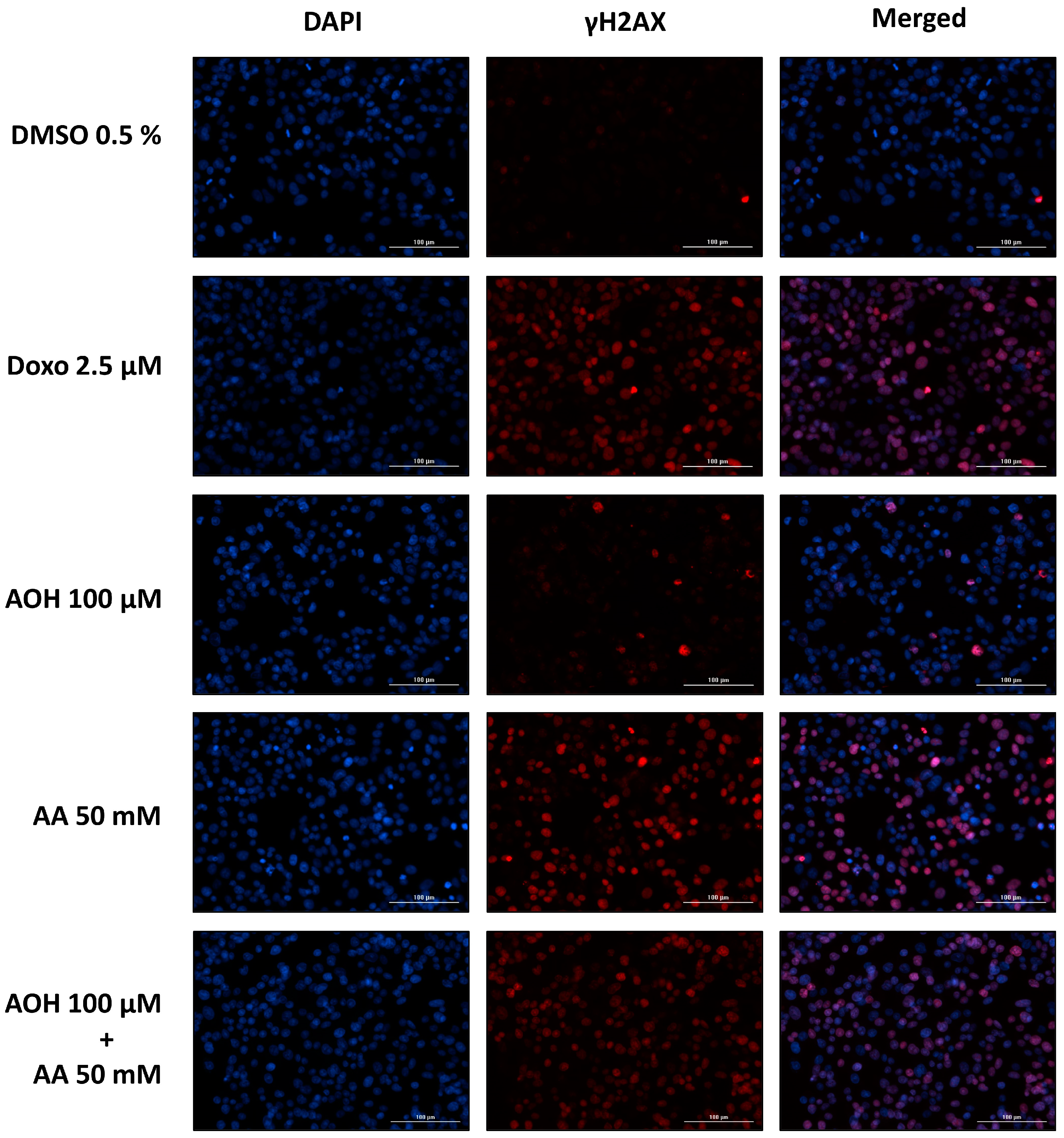
The genotoxic properties of the
Alternaria mycotoxin AOH in combination with AA were assessed by quantifying the phosphorylated histone γ-H2AX, whose production occurs following the induction of DNA double-stand breaks [
27]. As shown in
Figure 3, the exposure of cells to 0.5–50 mM AA resulted in a significant increase in γH2AX expression only at the highest concentration tested (50 mM), without differences between treatments with and without the liver S9 fraction. The induction of genotoxic effects by AA on HepG2 cells was previously reported by Jiang et al. [
28], who showed an increase in tail length and tail moment in the alkaline comet assay at concentrations far lower (2.5–20 mM) than those found to induce genotoxic effects in the present study (50 mM). This might be attributed to the fact that the alkaline comet assay assesses cumulative DNA damage resulting from both single-strand breaks (SSBs) and double-strand breaks (DSBs), while the phosphorylation of the 2AX histone occurs only in the presence of DSBs [
27]. It is important to note that DSBs are ordinarily present in smaller quantities than SSBs [
29], which further explains the onset in the present study of genotoxic effects at concentrations higher than those reported in the literature. Unlike AA, the exposure of cells to AOH resulted in a significant increase in γH2AX expression starting from a concentration of 10 µM. However, the increase was only slight and reached a plateau in the concentration range of 50–100 µM. The lack of a further increase in γH2AX expression in this concentration range is unlikely to be a consequence of a lower number of living cells due to cytotoxicity, since the γH2AX expression was assessed by quantifying the mean fluorescence signal per cell nucleus (and not per well). Furthermore, the co-exposure of cells to binary combinations of 50/100 µM AOH with 50 mM AA resulted in an increased γH2AX expression compared to that of the respective treatments with AOH alone. This confirms the non-involvement of the cytotoxic effects induced by AOH in the scarce increase in γH2AX expression during the single treatments with the mycotoxin. A possible explanation for this phenomenon might be the inhibition by the mycotoxin AOH of the kinases involved in the phosphorylation of the 2AX histone. In fact, AOH has been previously reported to inhibit casein kinase 2, probably by interacting with the ATP binding site of the enzyme [
30]. Due to the high conservation of ATP binding sites among kinases [
31], there is a plausible chance that AOH might also target the kinases involved in γH2AX formation. While additional focused investigations are essential to validate this assumption, this prospective mechanism could also explain the observed decrease in γH2AX expression observed when cells were exposed to the combination of “100 µM AOH + 50 mM AA” as opposed to 50 mM AA alone. In fact, on the one hand, the results from the γH2AX assay suggest a reduction in the genotoxic effects when cells are co-exposed to AOH and AA; however, on the other hand, this contradicts our expectations, given that both compounds individually induce genotoxic effects. Apart from the combination with the highest concentration of AOH, which resulted in antagonistic effects, no relevant combinatory effects were observed following the exposure of HepG2 cells to the other test conditions. In fact, the increases in γH2AX expression observed during the various co-treatments were comparable with those induced by the single treatments with the mycotoxin AOH (in co-treatments with 0.5, 5, and 10 mM AA) or the process contaminant AA (in co-treatments with 50 mM AA), depending on the compound showing the strongest genotoxic effects at the specific concentration tested.
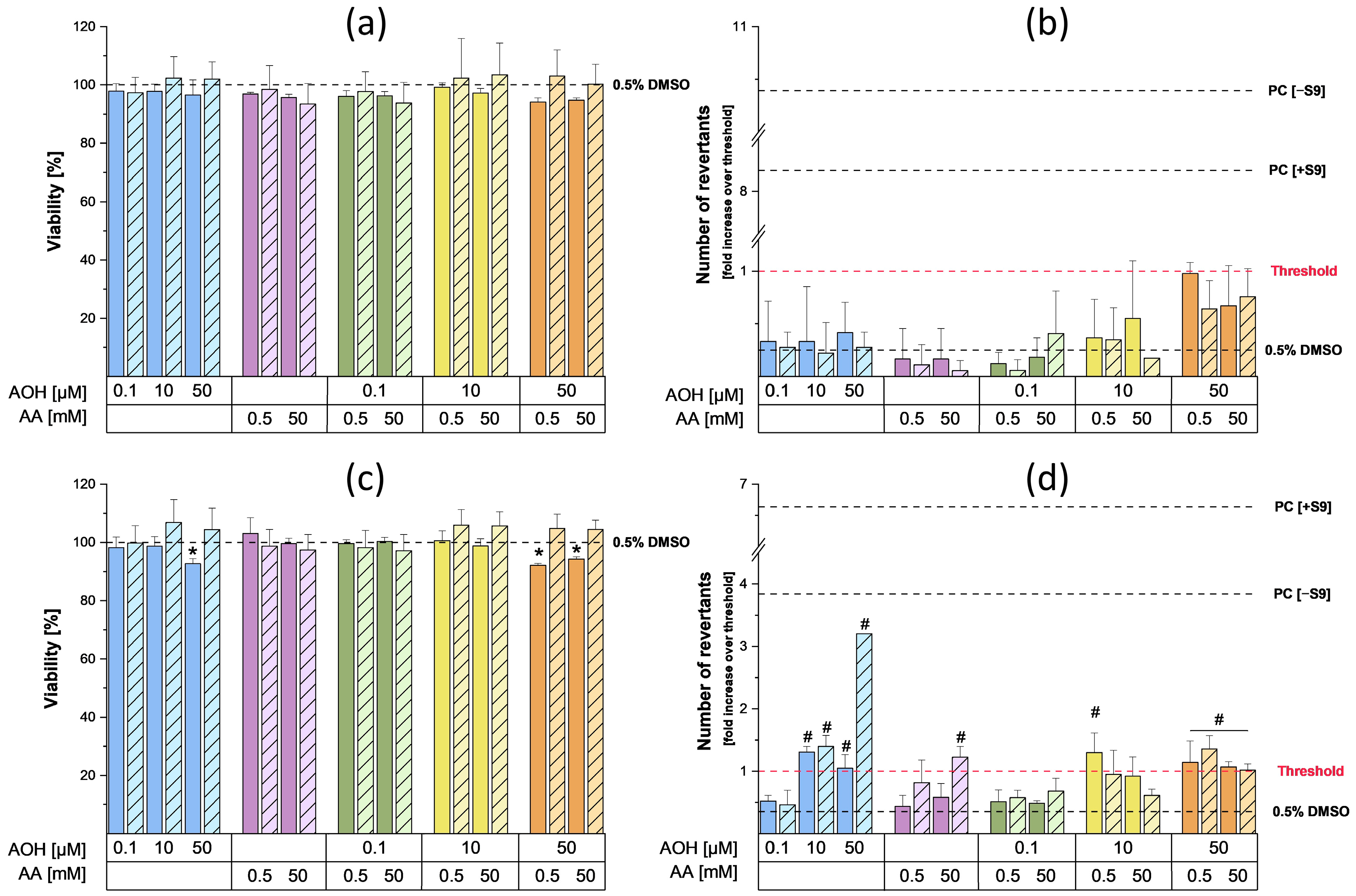
To assess the mutagenic properties of AOH and AA as single compounds and in combination, the Ames MPF test was conducted. For this purpose, two different strains of
Salmonella typhimurium, namely TA98 and TA100, were used to gain information about the type of mutations induced by the test compounds (TA98: frameshifts mutations; TA100: base pair substitutions) [
32]. The results of the preliminary cytotoxicity assessment performed on the strains did not show any cytotoxic effects following incubation with the various concentrations of AA. On the contrary, slight cytotoxic effects in the TA100 strain were observed in the presence of metabolic activation during the single and combined treatments with 50 µM AOH (the viability was always above 90%). As shown in
Figure 4b, the exposure of TA98 strain to 0.5 and 50 mM AA did not result in any increase in the number of revertants, while a slight but significant increase over the threshold was detected in the TA100 strain following exposure to 50 mM AA in the presence of metabolic activation (
Figure 4d). The increase in revertants observed after metabolic activation was probably a consequence of the metabolization of AA into its reactive metabolite glycidamide (GA), which is known to exert mutagenic effects in the
S. typhimurium TA100 strain [
33] and in several eukaryotic cell lines (formation of DNA adducts) [
18]. However, the currently available data on the mutagenic properties of AA on
S. typhimurium strains are often inconsistent. In fact, while Yang et al. showed weak positive results in both strains after metabolic activation [
34], other authors did not report any mutagenic effects [
35]. According to the IARC and FAO/WHO, the absence of clear mutagenic results in the Ames tests conducted on
S. typhimurium strains is probably a consequence of the limited availability or complete absence of a particular cytochrome P450 (CYP 450) isozyme (most likely CYP 2E1) in the S9 mix, which is responsible for metabolizing small hydrophilic compounds such as AA [
36]. In the present study, the usage of the Ames test in microplate format might also have contributed to our success in showing mutagenic effects in the TA100 strain compared to the previously reported studies in which the standard Ames test was performed. In fact, since the Ames MPF test requires the use of liquid media, bacteria are more in direct contact with the test compounds compared to the classic Ames test, which is performed on agar plates. Regarding the
Alternaria mycotoxin AOH, the exposure of the TA98 strain to 0.1, 10, and 50 µM AOH did not lead to any increase in the number of revertants compared to the solvent control (
Figure 4). On the contrary, mutagenic effects were observed in the TA100 strain starting from 10 µM, both in the presence and absence of metabolic activation. These results are in line with those reported by Schrader et al., who showed the inability of AOH to induce mutagenic effects in the TA98 strain, though it increased the number of TA100 revertants [
37]. In the present study, the exposure of the TA100 strain to AOH in the absence of metabolic activity only led to a slight increase in the number of revertants, while a more relevant and concentration-dependent mutagenic effect was observed during treatments in the presence of metabolic activation. To a much lesser extent, an enhancement in the mutagenic properties of AOH in the presence of the S9 fraction was also reported by Schrader et al. [
37]. This suggests that the metabolization of AOH could enhance its mutagenic properties. Interestingly, the mutagenic effect exerted by 50 µM AOH in the presence of metabolic activation was decreased by the co-incubation with both concentrations of AA. Even though further dedicated studies are needed to explain this phenomenon, this suppression might be a consequence of the reaction of AOH or AOH metabolites with AA or GA. This reaction could result in a reduced availability of the metabolite(s) responsible for the mutagenic effects induced by the mycotoxin. Finally, the possible activation of detoxification mechanisms by AOH, AA, or their respective metabolites at the concentrations tested cannot be excluded. Apart for the combinations with 50 µM AOH, which suggest the onset of antagonistic effects, no relevant combinatory effects were observed.
In summary, in the light of the results reported above, even though AOH and AA were shown to exert genotoxic and mutagenic effects when tested individually, their combination does not seem to result in the onset of combinatory effects. This was evident even at the concentrations tested, which significantly exceed those typically encountered through dietary exposure in vivo. Indeed, on the one hand, the lowest concentration tested for each of the test compounds might be potentially reached (e.g., at the intestinal level) if the extreme contamination levels reported in the literature are considered within the context of a varied diet; however, on the other hand, more realistic estimated exposure levels at the 95th percentile have been previously reported to range from 0.4 to 2.3 μg/kg bw/day for AA and 4.2 to 54.4 ng/kg bw/day for AOH [
16,
38]. These exposure levels would result in concentrations much lower than the ones tested in the present study, whose aim was to explore the possibility of the occurrence of combinatory effects in vitro. Nevertheless, it is important to note that, considering the complex composition of food, the potential onset of combined effects due to the co-presence of food bioactive compounds or other contaminants (e.g., mycotoxins from
Alternaria or other genera) cannot be entirely excluded.
5. Materials and Methods
5.1. Chemicals
Acrylamide (99%; for molecular biology), alternariol (96%; from Alternaria sp.), and doxorubicin hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA), while okadaic acid was acquired from Enzo Biochem Inc. (New York, NY, USA). For cell culture experiments, RPMI (Roswell Park Memorial Institute, Buffalo, NY, USA) 1640 medium, heat-inactivated fetal bovine serum, and penicillin/streptomycin (P/S) solution were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). For the cell viability assay, the CellTiter-Blue® Cell Viability reagent was purchased from Promega Corporation (Fitchburg, MA, USA). The 4′,6-diamidine-2′-phenylindoledihydrochloride (DAPI), Alexa Fluor ™ 633 F(ab’)2 fragment of goat anti-mouse IgG (H+L), bovine serum albumin (BSA; Standard Grade Powder, Fraction V), and S9 fraction (20 mg/mL from liver of Sprague Dawley rats), which were used for the γH2AX assay, were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Anti-phospho-Histone H2A.X (Ser139) Antibody (clone JBW301) and glucose-6-phosphate di-sodium salt were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA), while nicotinamide adenine dinucleotide phosphate (NADP) was purchased from Merck KGaA (Darmstadt, Germany).
5.2. Cell Culture Conditions
The CTB and γH2AX assays were performed by employing the human hepatocarcinoma HepG2 cell line (European Collection of Authenticated Cell Cultures; Salisbury, UK). Cells were maintained in RPMI 1640 medium supplemented with 10% (v/v) FBS and 1% penicillin (10,000 Units/mL)/streptomycin (10,000 µg/mL) solution. Cells were grown as a monolayer in a humidified incubator (37 °C, 5% CO2), passaged twice per week (70–80% confluency), and routinely tested for mycoplasma contamination.
5.3. In-Cell Western Assay of γ-H2AX
To assess the genotoxic properties of AOH (0.1, 10, 50, and 100 µM), AA (0.5, 5, 10, and 50 mM), and their respective binary combinations, the γH2AX assay was performed according to Ebmeyer et al. [
39], with some modifications. This assay is based on the quantification of the phosphorylated histone γ-H2AX, whose production occurs following the induction of double-strand breaks. All conditions were tested in the presence and absence of metabolic activation with rat liver S9 fraction. Briefly, 50,000 cells/well were seeded into 96-well plates (black plate, clear bottom; Corning Incorporated, Kennebunk, ME, USA) and allowed to attach for 24 h at 37 °C and 5% CO
2. Before cell treatment, an S9 mix consisting of 100 mM phosphate buffer (Na
2HPO
4/NaH
2PO
4), 8 mM MgCl
2, 33 mM KCl, 4 mM NADP, 5 mM glucose-6-phosphate, and 2 mg protein/mL of the S9 fraction was prepared for incubations requiring metabolic activation. The S9 mix was therefore diluted (1:5) with the various test/control media (RPMI + 1% P/S + controls/test substances) and pre-incubated at 37 °C and 5% CO
2 for 2 h. After 4 h incubation of cells with the various test conditions (with and without S9), cells were fixed by adding 50 µL of ice-cold methanol per well (20 min at 4 °C). Cells were washed three times with PBS-T (PBS + 0.1% Tween
® 20) and blocked (1% BSA in PBS-T) for 1 h at room temperature (subject to shaking). Afterwards, cells were washed once with PBS-T and incubated overnight (4 °C) with 100 µL/well of the primary antibody solution (Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301; 1:400 in blocking solution). Cells were washed three times with PBS-T and incubated for 1 h (at room temperature (RT), in the dark) with 100 µL/well of secondary antibody solution (Alexa Fluor™ 633 F(ab’)2 fragment of goat anti-mouse IgG (H+L); 1:500 in blocking solution). After washing with PBS-T, cell nuclei were stained by adding 50 µL of 3 µM DAPI solution (30 min at RT). Finally, cells were washed three times with PBS-T and plates were stored at 4 °C until the time of analysis. Picture acquisition (two optical fields/well) and quantification of the γH2AX fluorescence signals were performed by using a Lionheart FX automated microscope equipped with the software Gen5 (ver. 3.08; BioTek Instruments Inc., Winooski, VT, USA). The corresponding wavelengths for the detection of the fluorescence signals were 380/460 nm (excitation/emission) for DAPI and 620/655 nm (excitation/emission) for the γ-H2AX. The mean value of the γH2AX signal intensity per cell was calculated and values were normalized to the positive control (2.5 µM doxorubicin). As controls, 0.5% DMSO, 2.5 µM doxorubicin (for treatments without S9 fraction), and 0.1 µM okadaic acid (for treatments with S9 fraction) were used. The final DMSO concentration was always 0.5%. All test conditions were tested in technical triplicates and with 3 independent experiments.
5.4. CellTiter-Blue® Assay
The CellTiter-Blue
® assay was applied to assess the effects of AOH, AA, and their respective combinations on the viability of HepG2 cells. After the cells were incubated for 4 h with the various test conditions (see
Section 5.3 for cell seeding and treatment), the test media were removed and replaced with 100 µL/well of the 1× CTB solution (1:10 dilution of 10× CTB solution with phenol-red free DMEM). After 40 min of incubation at 37 °C and 5% CO
2, the supernatants were transferred into a black 96-well plate and the fluorescence was measured at 590 nm by means of a microplate reader (Cytation 3 imaging reader; BioTek, USA). All test conditions were tested in technical triplicates and with three independent experiments. Results were expressed as mean fluorescence value of three biological replicates and normalized to the solvent control (0.5% DMSO; set to 100%).
5.5. Ames Microplate Format (MPF™) Assay
To assess the mutagenic properties of AOH (0.1, 10, and 50 µM), AA (0.5 and 50 mM), and their respective binary combinations, a commercial Ames kit with aroclor-induced rat liver S9 fraction was employed (Ames MPF 98/100 semisolid kit + Ames MPF S9 Cofactor Kit; Xenometrix® AG, Allschwil, Switzerland). In particular, the test was conducted following the provider’s protocol and by employing the S. typhimurium strains TA98 (sensitive to frameshift mutations) and TA100 (base pair substitution). Briefly, overnight cultures of bacteria were used only after reaching an optical density at 600 nm (OD600 value) ≥ 2. For incubations without S9 fraction, overnight cultures were diluted to 1:10 (for TA98) or 1:20 (TA100) with exposure media containing the various test chemicals. For incubations with S9 fraction, an S9 mix containing buffer salts, glucose-6-phosphate, NADP, and S9 fraction was prepared according to the provider’s protocol and diluted 1:6 with exposure media containing booster solution (final concentration: 0.16%) and the various test chemicals. For treatments with TA98 and TA100 strains in absence of S9 fraction, a mixture of 2 µg/mL 2-nitrofluorene and 0.1 µg/mL 4-nitroquinoline oxide was used as a positive control. For treatments of TA98 and TA100 in the presence of metabolic activation, 1 and 2.5 µg/mL 2-aminoanthracene were used as positive controls. DMSO (4%) was used as a solvent control (with the same % in all test conditions). Bacterial suspensions (250 µL/well in 24-well plates) were therefore incubated with the various test conditions for 90 min at 37 °C while shaken. At the end of the incubation time, 2.8 mL of indicator medium that lacks histidine was added to each well. Afterwards, 50 µL of each bacterial suspension (treated and diluted in indicator medium) was transferred into 48 wells of a 384-well plate, which was subsequently placed into a resealable plastic bag and incubated for 48 h at 37 °C (without shaking). The plates were scored by counting the wells that turned yellow. Results were expressed as fold increase over threshold, which was calculated as two times the sum of the mean value of the solvent control plus one standard deviation (in accordance with the provider’s instructions). The test was considered positive when the fold induction was higher than the threshold.
To assess the impact of single and combined treatments on the viability of bacteria, a cell viability test (in exposure medium) was performed on bacterial strains exposed to the various test conditions (for 90 min) by measuring the optical density at 600 nm. Furthermore, to avoid misinterpretation of the results due to poor solubility of the compounds, the presence of possible crystals in the wells was assessed by using an inverted microscope.
The concentration range chosen for AOH and AA aligned with OECD guideline n. 471, which delineates the evaluation of the mutagenic properties of chemicals via bacterial reverse mutation tests [
40]. Specifically, the highest concentrations tested in this study were those that did not interfere with plate scoring due to precipitation of the compounds.
5.6. Statistical Analyses
The statistical analyses were conducted using Origin Pro 2022. Significant differences between single and combined treatments as well as between treatments with and without metabolic activation were evaluated by applying the Student’s t-test. One-way ANOVA with Fisher’s LSD post-hoc test was applied to assess the differences between the various treatments and the respective solvent or positive control.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}