Chemical Synthesis of a Functional Fluorescent-Tagged α-Bungarotoxin

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
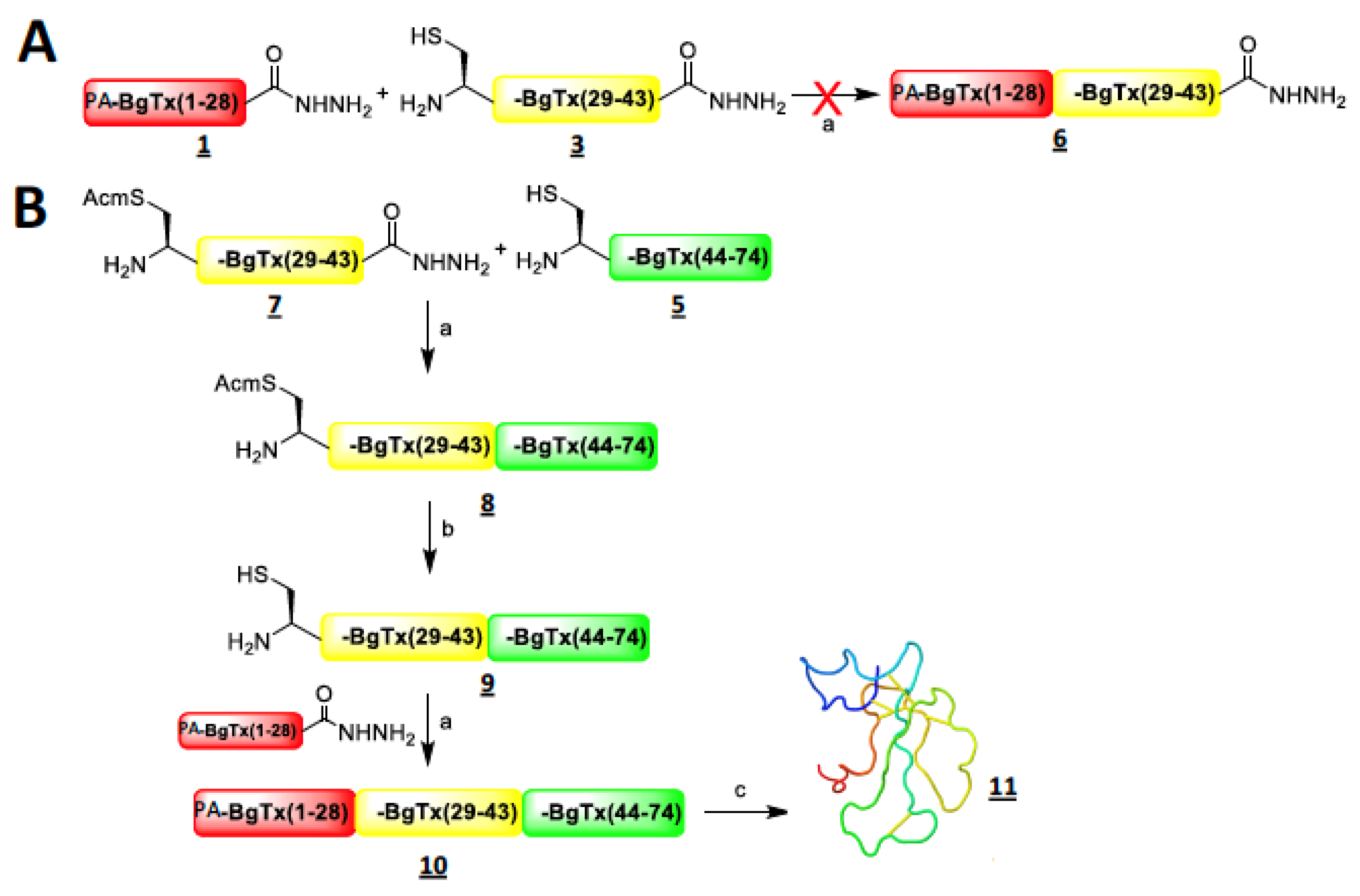
2.1. Strategy of Synthesis and Chemical Production of Intermediate Fragments of α-Bungarotoxin
2.2. Chemical Synthesis of Full-Length α-Bungarotoxin
2.3. Coupling of a Fluorochrome on PA-α-BgTx by Click Chemistry
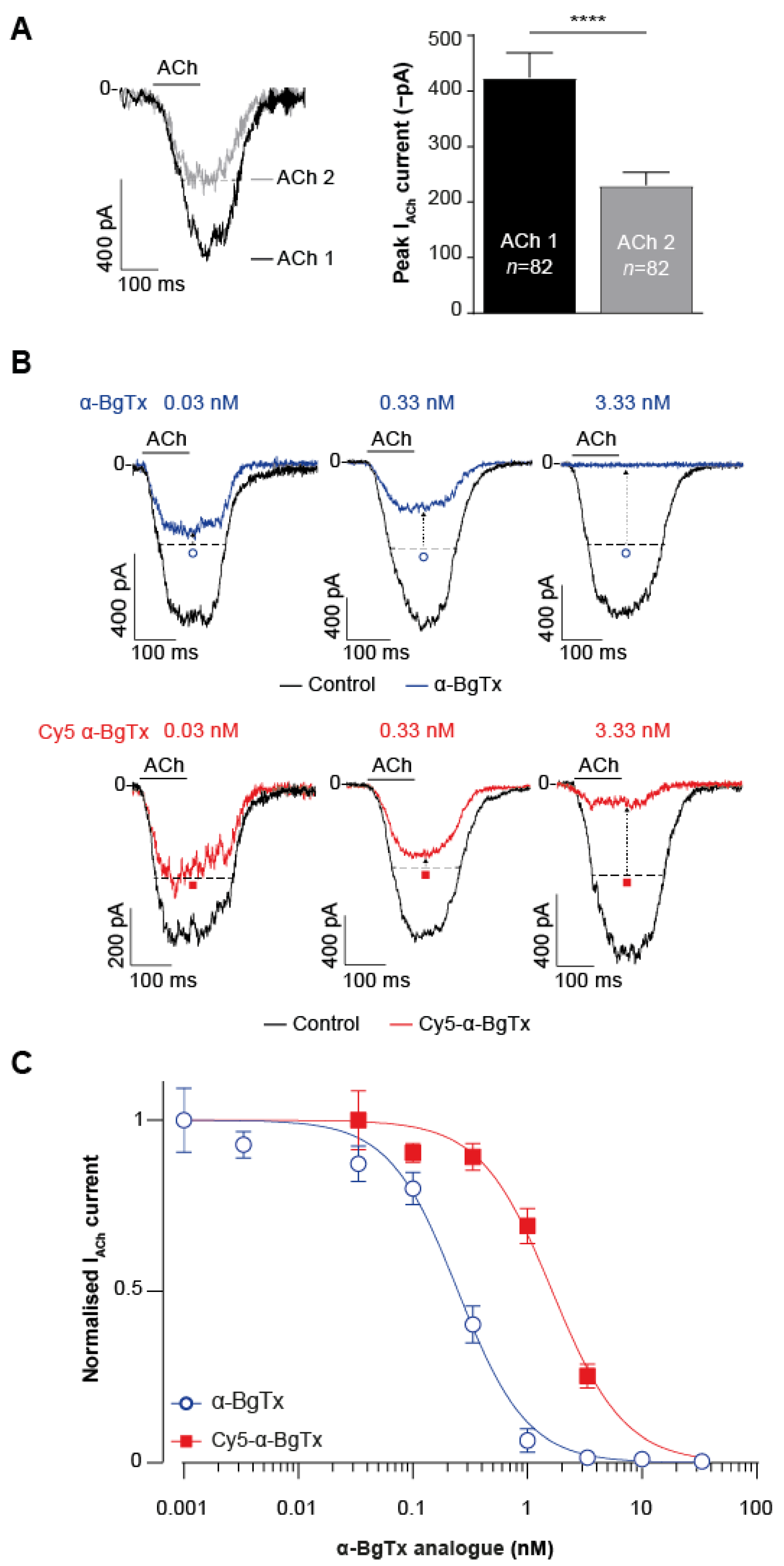
2.4. High-Throughput Evaluation of ACh and Cy5-α-Bungarotoxin Effects on the TE671 Cell Line
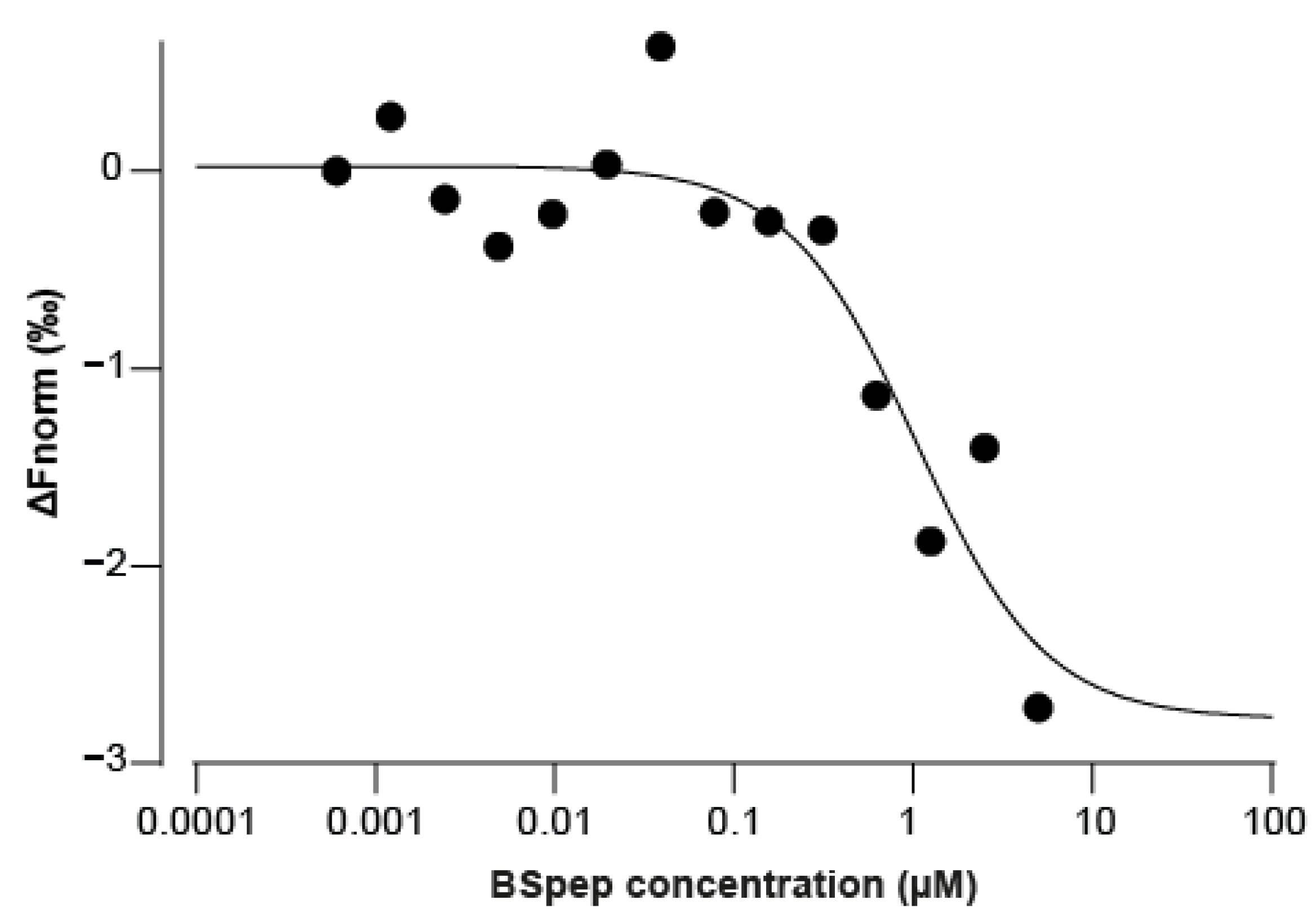
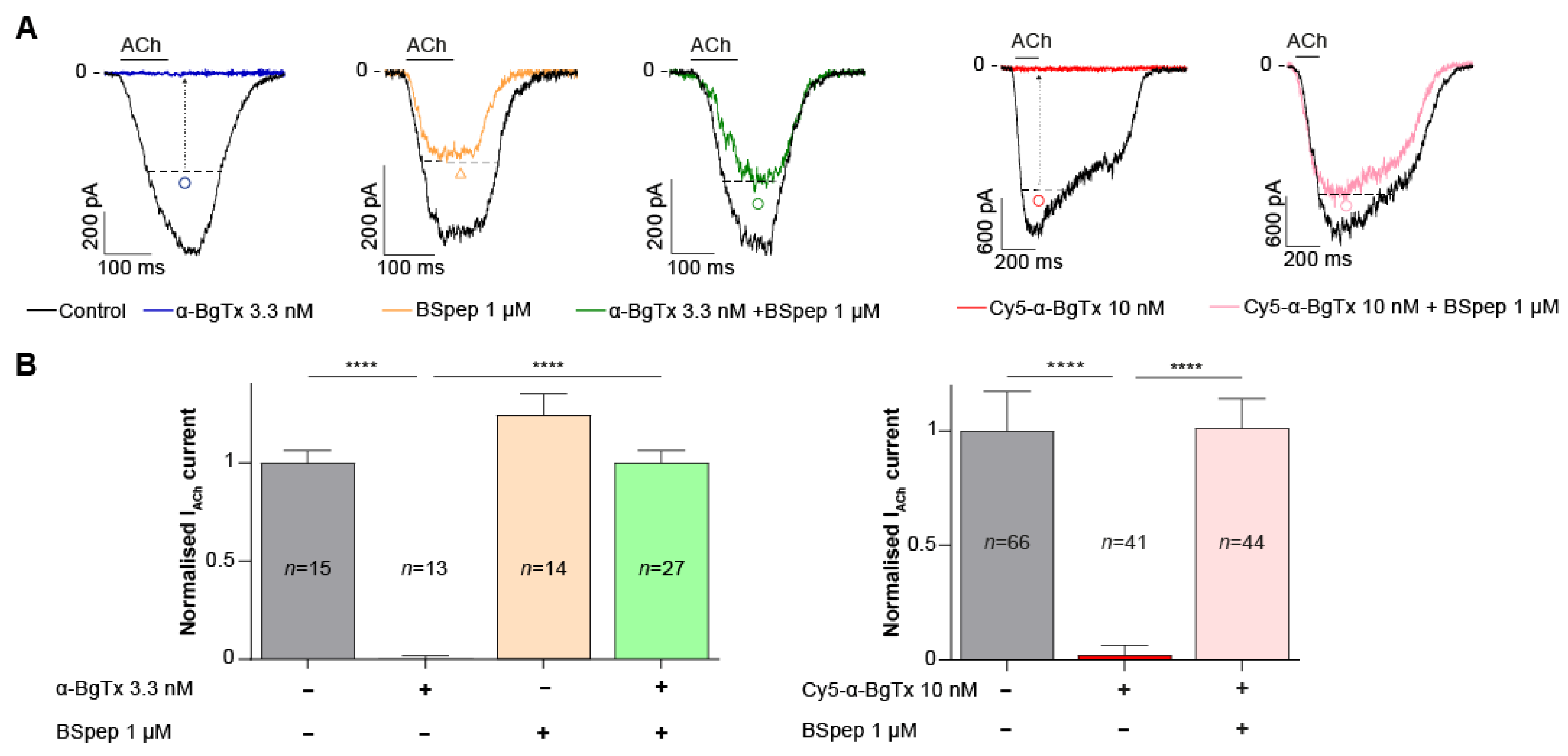
2.5. Neutralisation of α-BgTx-Mediated Inhibition of ACh Response by a Binding Site-Mimicking Peptide
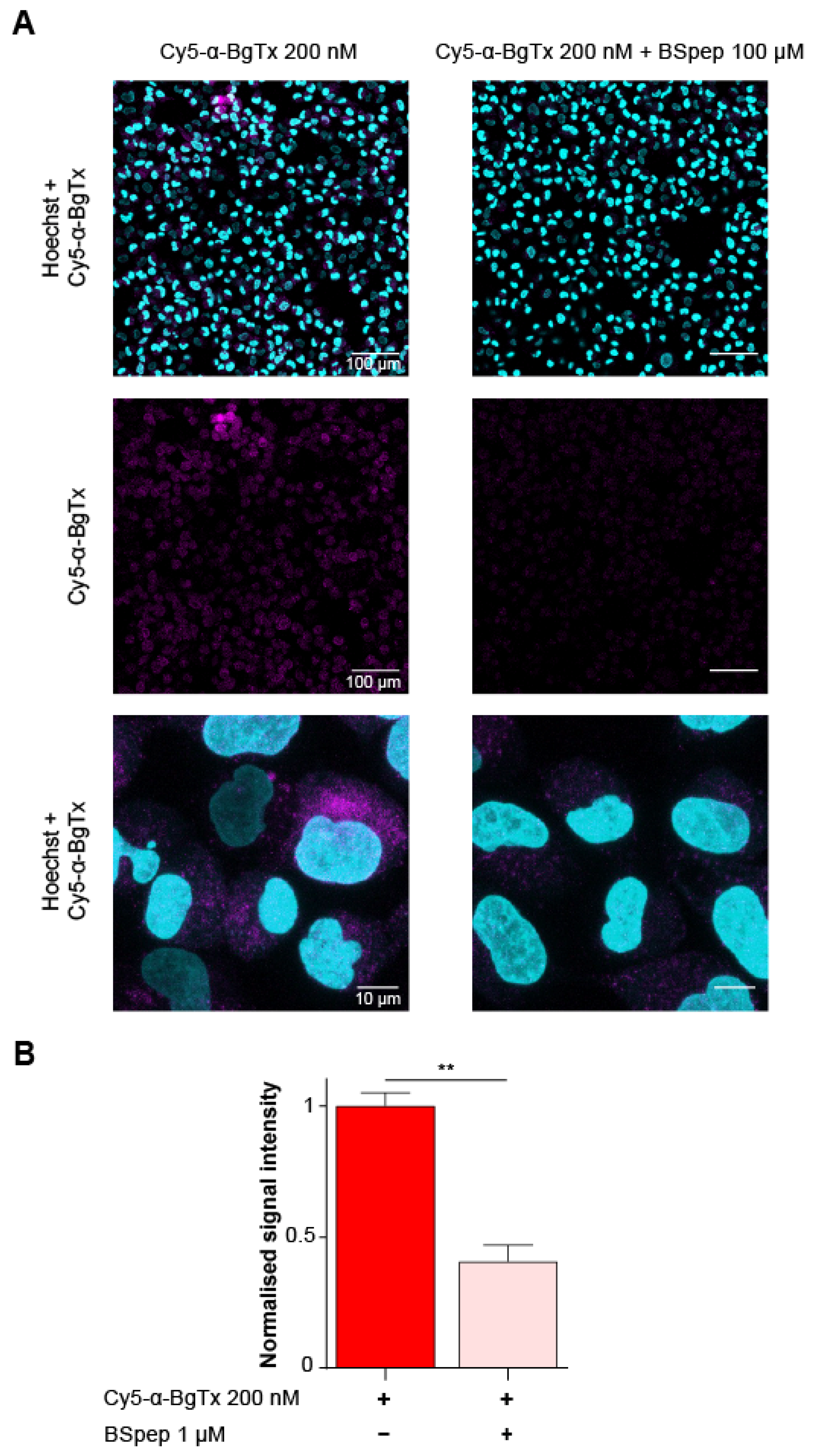
2.6. Characterisation of the Cellular Distribution of Muscle-Type mAChR with Fluorescent Cy5-α-BgTx
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Resin Loading for Solid-Phase Peptide Syntheses
4.3. Solid-Phase Peptide Syntheses
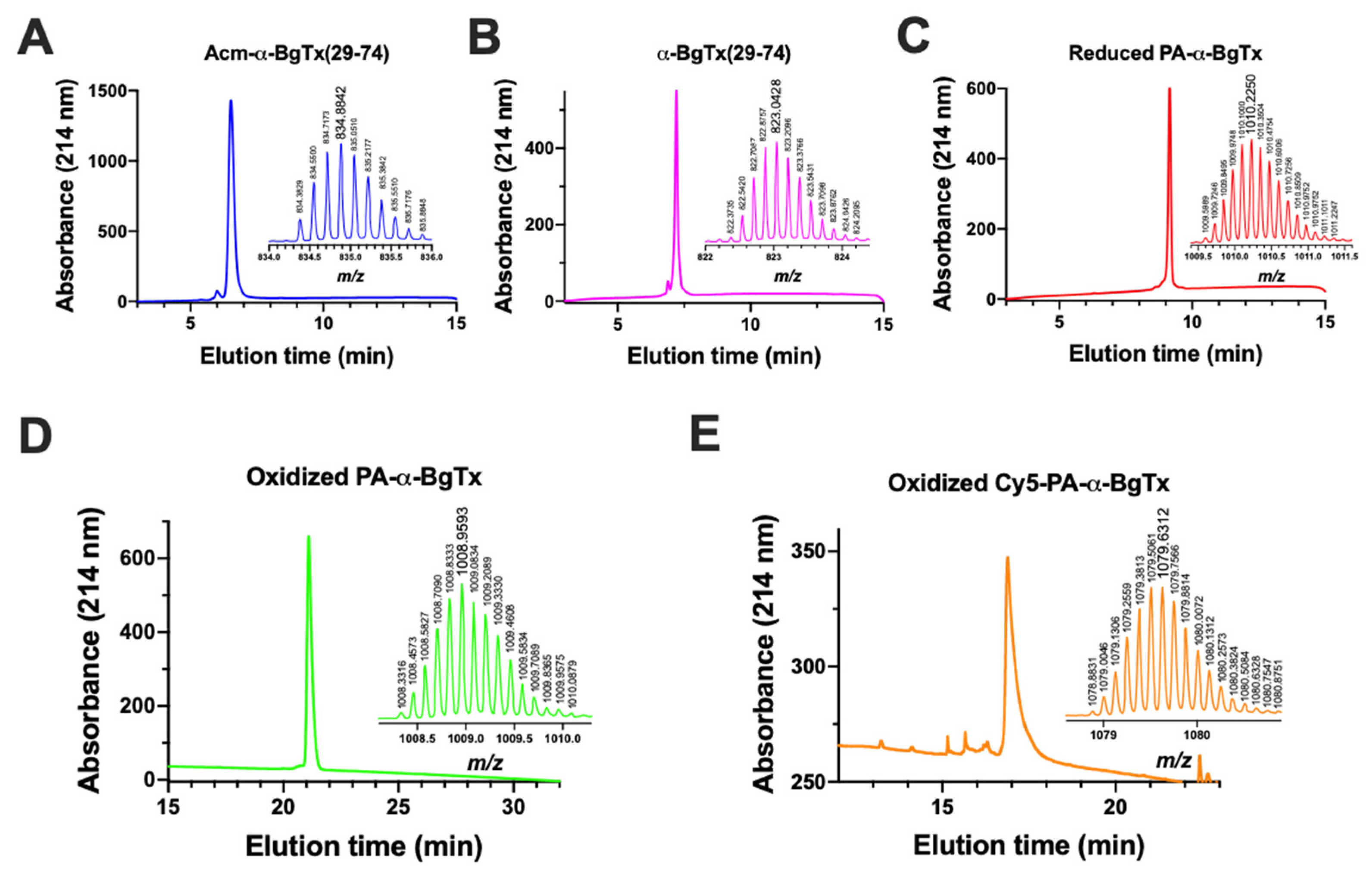
4.4. HPLC and Mass Spectrometry Analyses
4.5. Reduced PA-α-BgTx Synthesis by Native Chemical Ligation
4.6. Folding of Reduced PA-α-BgTx
4.7. Coupling of Cy5 on PA-α-BgTx
4.8. Cell Culture
4.9. Ligand Binding Studies
4.10. Automated Patch-Clamp Recordings
4.11. Cell Imaging by Confocal Microscopy
4.12. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mebs, D.; Narita, K.; Iwanaga, S.; Samejima, Y.; Lee, C.Y. Purification, properties and amino acid sequence of alpha-bungarotoxin from the venom of Bungarus multicinctus. Hoppe-Seyler’s Z. Für Physiol. Chem. 1972, 353, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Kasai, M.; Lee, C.Y. Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc. Natl. Acad. Sci. USA 1970, 67, 1241–1247. [Google Scholar] [CrossRef]
- daCosta, C.J.; Free, C.R.; Sine, S.M. Stoichiometry for alpha-bungarotoxin block of alpha7 acetylcholine receptors. Nat. Commun. 2015, 6, 8057. [Google Scholar] [CrossRef] [PubMed]
- Dellisanti, C.D.; Yao, Y.; Stroud, J.C.; Wang, Z.Z.; Chen, L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 A resolution. Nat. Neurosci. 2007, 10, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Swinburne, I.A.; Mosaliganti, K.R.; Green, A.A.; Megason, S.G. Improved Long-Term Imaging of Embryos with Genetically Encoded alpha-Bungarotoxin. PLoS ONE 2015, 10, e0134005. [Google Scholar] [CrossRef]
- Paulo, J.A.; Hawrot, E. A radioisotope label-free alpha-bungarotoxin-binding assay using BIAcore sensor chip technology for real-time analysis. Anal. Biochem. 2009, 389, 86–88. [Google Scholar] [CrossRef][Green Version]
- Berg, D.K.; Kelly, R.B.; Sargent, P.B.; Williamson, P.; Hall, Z.W. Binding of -bungarotoxin to acetylcholine receptors in mammalian muscle (snake venom-denervated muscle-neonatal muscle-rat diaphragm-SDS-polyacrylamide gel electrophoresis). Proc. Natl. Acad. Sci. USA 1972, 69, 147–151. [Google Scholar] [CrossRef]
- Fambrough, D.M.; Hartzell, H.C. Acetylcholine receptors: Number and distribution at neuromuscular junctions in rat diaphragm. Science 1972, 176, 189–191. [Google Scholar] [CrossRef]
- Raftery, M.A. Isolation of acetylcholine receptor—α-Bungarotoxin complexes from Torpedo californica electroplax. Arch. Biochem. Biophys. 1973, 154, 270–276. [Google Scholar] [CrossRef]
- Chazotte, B. Labeling Acetylcholine Receptors in Live Cells Using Rhodamine {alpha}-Bungarotoxin for Imaging. CSH Protoc. 2008, 2008, pdb-prot4928. [Google Scholar] [CrossRef]
- Anderson, M.J.; Cohen, M.W. Fluorescent staining of acetylcholine receptors in vertebrate skeletal muscle. J. Physiol. 1974, 237, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Fertuck, H.C.; Salpeter, M.M. Localization of acetylcholine receptor by 125I-labeled alpha-bungarotoxin binding at mouse motor endplates. Proc. Natl. Acad. Sci. USA 1974, 71, 1376–1378. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, T.; Chen, B.; Qi, Y.; Zhang, P.; Zhu, D.; Yin, X.; Jiang, B. In vivo injection of alpha-bungarotoxin to improve the efficiency of motor endplate labeling. Brain Behav. 2016, 6, e00468. [Google Scholar] [CrossRef]
- Vogel, Z.; Towbin, M.; Daniels, M.P. Alpha-bungarotoxin-horseradish peroxidase conjugate: Preparation, properties and utilization for the histochemical detection of acetylcholine receptors. J. Histochem. Cytochem. 1979, 27, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.W.; Barik, J.; O’Neill, M.J.; Wonnacott, S. Alpha bungarotoxin-1.4 nm gold: A novel conjugate for visualising the precise subcellular distribution of alpha 7* nicotinic acetylcholine receptors. J. Neurosci. Methods 2004, 134, 65–74. [Google Scholar] [CrossRef]
- Harel, M.; Kasher, R.; Nicolas, A.; Guss, J.M.; Balass, M.; Fridkin, M.; Smit, A.B.; Brejc, K.; Sixma, T.K.; Katchalski-Katzir, E.; et al. The binding site of acetylcholine receptor as visualized in the X-Ray structure of a complex between alpha-bungarotoxin and a mimotope peptide. Neuron 2001, 32, 265–275. [Google Scholar] [CrossRef]
- Bernini, A.; Ciutti, A.; Spiga, O.; Scarselli, M.; Klein, S.; Vannetti, S.; Bracci, L.; Lozzi, L.; Lelli, B.; Falciani, C.; et al. NMR and MD studies on the interaction between ligand peptides and alpha-bungarotoxin. J. Mol. Biol. 2004, 339, 1169–1177. [Google Scholar] [CrossRef]
- Bracci, L.; Lozzi, L.; Pini, A.; Lelli, B.; Falciani, C.; Niccolai, N.; Bernini, A.; Spreafico, A.; Soldani, P.; Neri, P. A branched peptide mimotope of the nicotinic receptor binding site is a potent synthetic antidote against the snake neurotoxin alpha-bungarotoxin. Biochemistry 2002, 41, 10194–10199. [Google Scholar] [CrossRef] [PubMed]
- Tabor, G.T.; Park, J.M.; Murphy, J.G.; Hu, J.H.; Hoffman, D.A. A novel bungarotoxin binding site-tagged construct reveals MAPK-dependent Kv4.2 trafficking. Mol. Cell Neurosci. 2019, 98, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Watschinger, K.; Horak, S.B.; Schulze, K.; Obermair, G.J.; Wild, C.; Koschak, A.; Sinnegger-Brauns, M.J.; Tampe, R.; Striessnig, J. Functional properties and modulation of extracellular epitope-tagged Ca(V)2.1 voltage-gated calcium channels. Channels 2008, 2, 461–473. [Google Scholar] [CrossRef] [PubMed]
- McCann, C.M.; Bareyre, F.M.; Lichtman, J.W.; Sanes, J.R. Peptide tags for labeling membrane proteins in live cells with multiple fluorophores. Biotechniques 2005, 38, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Hannan, S.; Wilkins, M.E.; Thomas, P.; Smart, T.G. Tracking cell surface mobility of GPCRs using alpha-bungarotoxin-linked fluorophores. Methods Enzymol. 2013, 521, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.A.; Luo, G.; Davis, M.I.; Hales, T.G.; Lovinger, D.M. Fluorophore assisted light inactivation (FALI) of recombinant 5-HT(3)A receptor constitutive internalization and function. Mol. Cell Neurosci. 2011, 47, 79–92. [Google Scholar] [CrossRef][Green Version]
- Guo, J.; Chen, H.; Puhl, H.L., 3rd; Ikeda, S.R. Fluorophore-assisted light inactivation produces both targeted and collateral effects on N-type calcium channel modulation in rat sympathetic neurons. J. Physiol. 2006, 576, 477–492. [Google Scholar] [CrossRef]
- Sekine-Aizawa, Y.; Huganir, R.L. Imaging of receptor trafficking by using alpha-bungarotoxin-binding-site-tagged receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 17114–17119. [Google Scholar] [CrossRef] [PubMed]
- Caffery, P.M.; Krishnaswamy, A.; Sanders, T.; Liu, J.; Hartlaub, H.; Klysik, J.; Cooper, E.; Hawrot, E. Engineering neuronal nicotinic acetylcholine receptors with functional sensitivity to alpha-bungarotoxin: A novel alpha3-knock-in mouse. Eur. J. Neurosci. 2009, 30, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T.; Hawrot, E. A novel pharmatope tag inserted into the beta4 subunit confers allosteric modulation to neuronal nicotinic receptors. J. Biol. Chem. 2004, 279, 51460–51465. [Google Scholar] [CrossRef]
- Xu, J.; Li, J.; Wu, X.; Song, C.; Lin, Y.; Shen, Y.; Ye, W.; Sun, C.; Wang, X.; Li, Z.; et al. Expression and refolding of bioactive alpha-bungarotoxin V31 in E. coli. Protein Expr. Purif. 2015, 110, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Qi, G.; Jun, L.; Ying, L.; Yong, Z.; Dongliang, H.; Changlin, T. Total synthesis of snake toxin alpha-bungarotoxin and its analogues by hydrazide-based native chemical ligation. Chin. Chem. Lett. 2018, 29, 1139–1142. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Cargoet, M.; Melnyk, O. A statistical view of protein chemical synthesis using NCL and extended methodologies. Bioorg. Med. Chem. 2017, 25, 4938–4945. [Google Scholar] [CrossRef]
- Fang, G.M.; Li, Y.M.; Shen, F.; Huang, Y.C.; Li, J.B.; Lin, Y.; Cui, H.K.; Liu, L. Protein chemical synthesis by ligation of peptide hydrazides. Angew. Chem. Int. Ed. Engl. 2011, 50, 7645–7649. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, T.M.; Griffin, J.H.; Dawson, P.E. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. USA 1999, 96, 10068–10073. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Canosa, J.B.; Dawson, P.E. An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew. Chem. Int. Ed. Engl. 2008, 47, 6851–6855. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Fang, G.M.; He, Y.; Qu, D.L.; Yu, M.; Hong, Z.Y.; Liu, L. Peptide o-aminoanilides as crypto-thioesters for protein chemical synthesis. Angew. Chem. Int. Ed. Engl. 2015, 54, 2194–2198. [Google Scholar] [CrossRef]
- Blanco-Canosa, J.B.; Nardone, B.; Albericio, F.; Dawson, P.E. Chemical Protein Synthesis Using a Second-Generation N-Acylurea Linker for the Preparation of Peptide-Thioester Precursors. J. Am. Chem. Soc. 2015, 137, 7197–7209. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoet, M.; Monbaliu, J.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef] [PubMed]
- Laps, S.; Sun, H.; Kamnesky, G.; Brik, A. Palladium-Mediated Direct Disulfide Bond Formation in Proteins Containing S-Acetamidomethyl-cysteine under Aqueous Conditions. Angew. Chem. Int. Ed. Engl. 2019, 58, 5729–5733. [Google Scholar] [CrossRef] [PubMed]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. Engl. 2009, 48, 9879–9883. [Google Scholar] [CrossRef]
- Stratton, M.R.; Darling, J.; Pilkington, G.J.; Lantos, P.L.; Reeves, B.R.; Cooper, C.S. Characterization of the human cell line TE671. Carcinogenesis 1989, 10, 899–905. [Google Scholar] [CrossRef]
- Schoepfer, R.; Luther, M.; Lindstrom, J. The human medulloblastoma cell line TE671 expresses a muscle-like acetylcholine receptor. Cloning of the alpha-subunit cDNA. FEBS Lett. 1988, 226, 235–240. [Google Scholar] [CrossRef]
- Shao, Z.; Mellor, I.R.; Brierley, M.J.; Harris, J.; Usherwood, P.N. Potentiation and inhibition of nicotinic acetylcholine receptors by spermine in the TE671 human muscle cell line. J. Pharmacol. Exp. Ther. 1998, 286, 1269–1276. [Google Scholar] [PubMed]
- Dudel, J.; Franke, C.; Hatt, H. Rapid activation and desensitization of transmitter-liganded receptor channels by pulses of agonists. Ion Channels 1992, 3, 207–260. [Google Scholar] [CrossRef] [PubMed]
- Franke, C.; Koltgen, D.; Hatt, H.; Dudel, J. Activation and desensitization of embryonic-like receptor channels in mouse muscle by acetylcholine concentration steps. J. Physiol. 1992, 451, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Taiwe, G.S.; Montnach, J.; Nicolas, S.; De Waard, S.; Fiore, E.; Peyrin, E.; El-Aziz, T.M.A.; Amar, M.; Molgo, J.; Ronjat, M.; et al. Aptamer Efficacies for In Vitro and In Vivo Modulation of alphaC-Conotoxin PrXA Pharmacology. Molecules 2019, 24, 229. [Google Scholar] [CrossRef] [PubMed]
- Syapin, P.J.; Salvaterra, P.M.; Engelhardt, J.K. Neuronal-like features of TE671 cells: Presence of a functional nicotinic cholinergic receptor. Brain Res. 1982, 231, 365–377. [Google Scholar] [CrossRef]
- Cetin, H.; Liu, W.; Cheung, J.; Cossins, J.; Vanhaesebrouck, A.; Maxwell, S.; Vincent, A.; Beeson, D.; Webster, R. Rapsyn facilitates recovery from desensitization in fetal and adult acetylcholine receptors expressed in a muscle cell line. J. Physiol. 2019, 597, 3713–3725. [Google Scholar] [CrossRef]
- Conroy, W.G.; Saedi, M.S.; Lindstrom, J. TE671 cells express an abundance of a partially mature acetylcholine receptor alpha subunit which has characteristics of an assembly intermediate. J. Biol. Chem. 1990, 265, 21642–21651. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Montnach, J.; De Waard, S.; Nicolas, S.; Burel, S.; Osorio, N.; Zoukimian, C.; Mantegazza, M.; Boukaiba, R.; Beroud, R.; Partiseti, M.; et al. Fluorescent- and tagged-protoxin II peptides: Potent markers of the Nav 1.7 channel pain target. Br. J. Pharmacol. 2021, 178, 2632–2650. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brun, O.; Zoukimian, C.; Oliveira-Mendes, B.; Montnach, J.; Lauzier, B.; Ronjat, M.; Béroud, R.; Lesage, F.; Boturyn, D.; De Waard, M. Chemical Synthesis of a Functional Fluorescent-Tagged α-Bungarotoxin. Toxins 2022, 14, 79. https://doi.org/10.3390/toxins14020079
Brun O, Zoukimian C, Oliveira-Mendes B, Montnach J, Lauzier B, Ronjat M, Béroud R, Lesage F, Boturyn D, De Waard M. Chemical Synthesis of a Functional Fluorescent-Tagged α-Bungarotoxin. Toxins. 2022; 14(2):79. https://doi.org/10.3390/toxins14020079
Chicago/Turabian StyleBrun, Oliver, Claude Zoukimian, Barbara Oliveira-Mendes, Jérôme Montnach, Benjamin Lauzier, Michel Ronjat, Rémy Béroud, Frédéric Lesage, Didier Boturyn, and Michel De Waard. 2022. "Chemical Synthesis of a Functional Fluorescent-Tagged α-Bungarotoxin" Toxins 14, no. 2: 79. https://doi.org/10.3390/toxins14020079
APA StyleBrun, O., Zoukimian, C., Oliveira-Mendes, B., Montnach, J., Lauzier, B., Ronjat, M., Béroud, R., Lesage, F., Boturyn, D., & De Waard, M. (2022). Chemical Synthesis of a Functional Fluorescent-Tagged α-Bungarotoxin. Toxins, 14(2), 79. https://doi.org/10.3390/toxins14020079