Variability in the Spatial Structure of the Central Loop in Cobra Cytotoxins Revealed by X-ray Analysis and Molecular Modeling

 ,
, 
 and
and
Abstract
:
1. Introduction
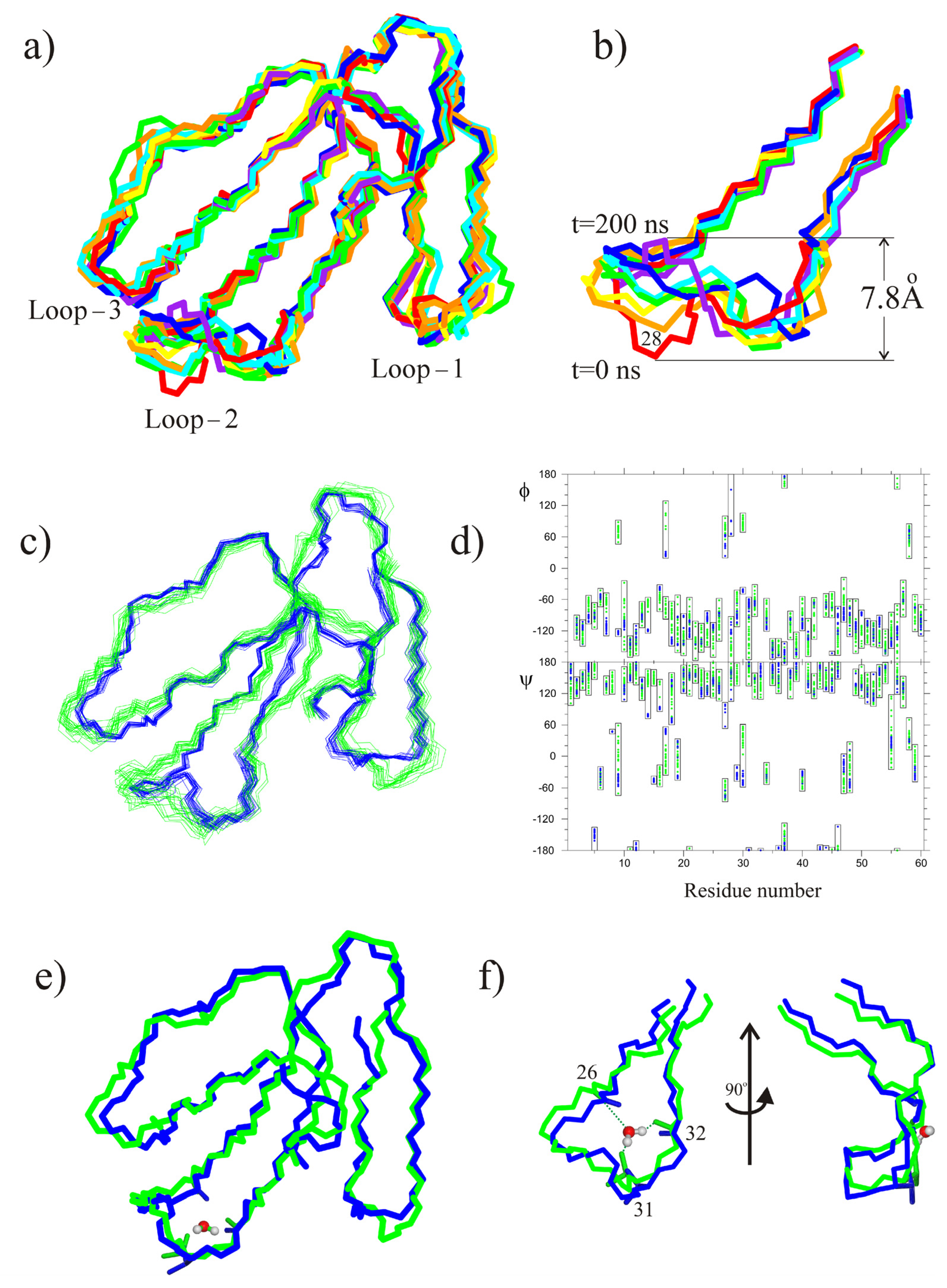
2. Results
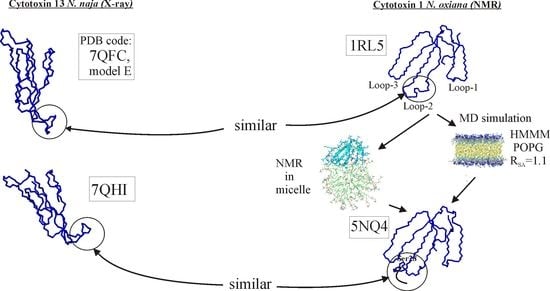
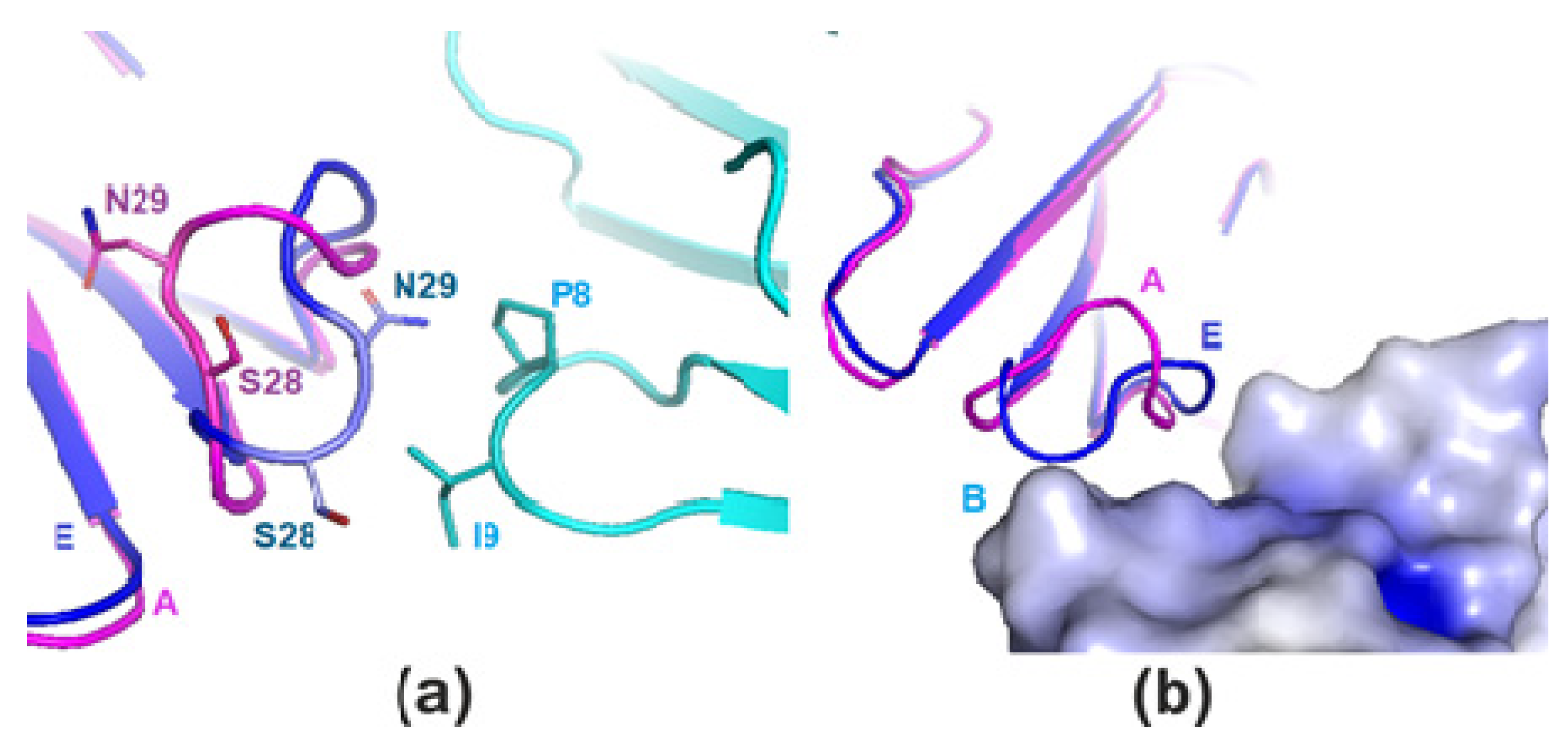
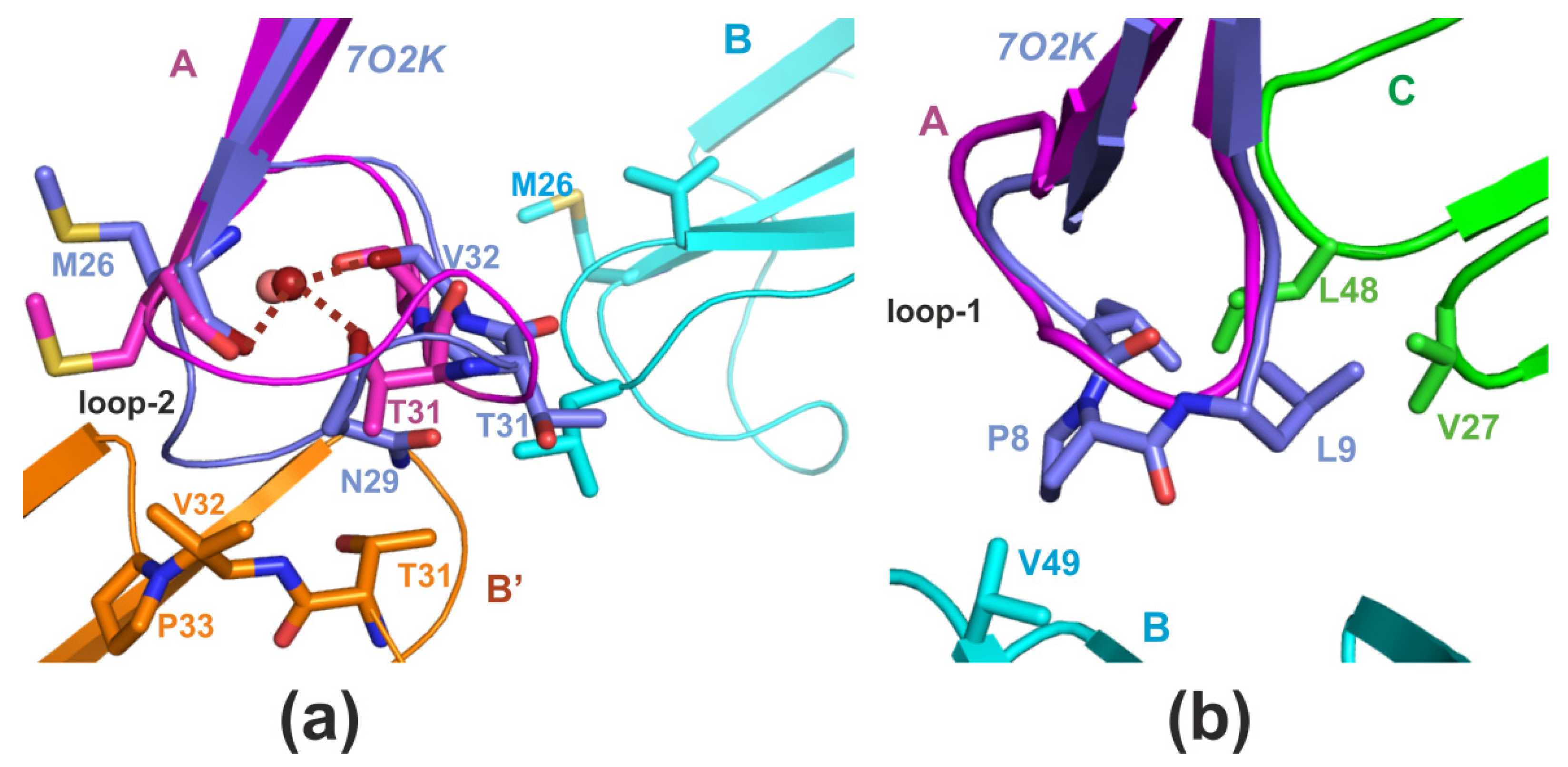
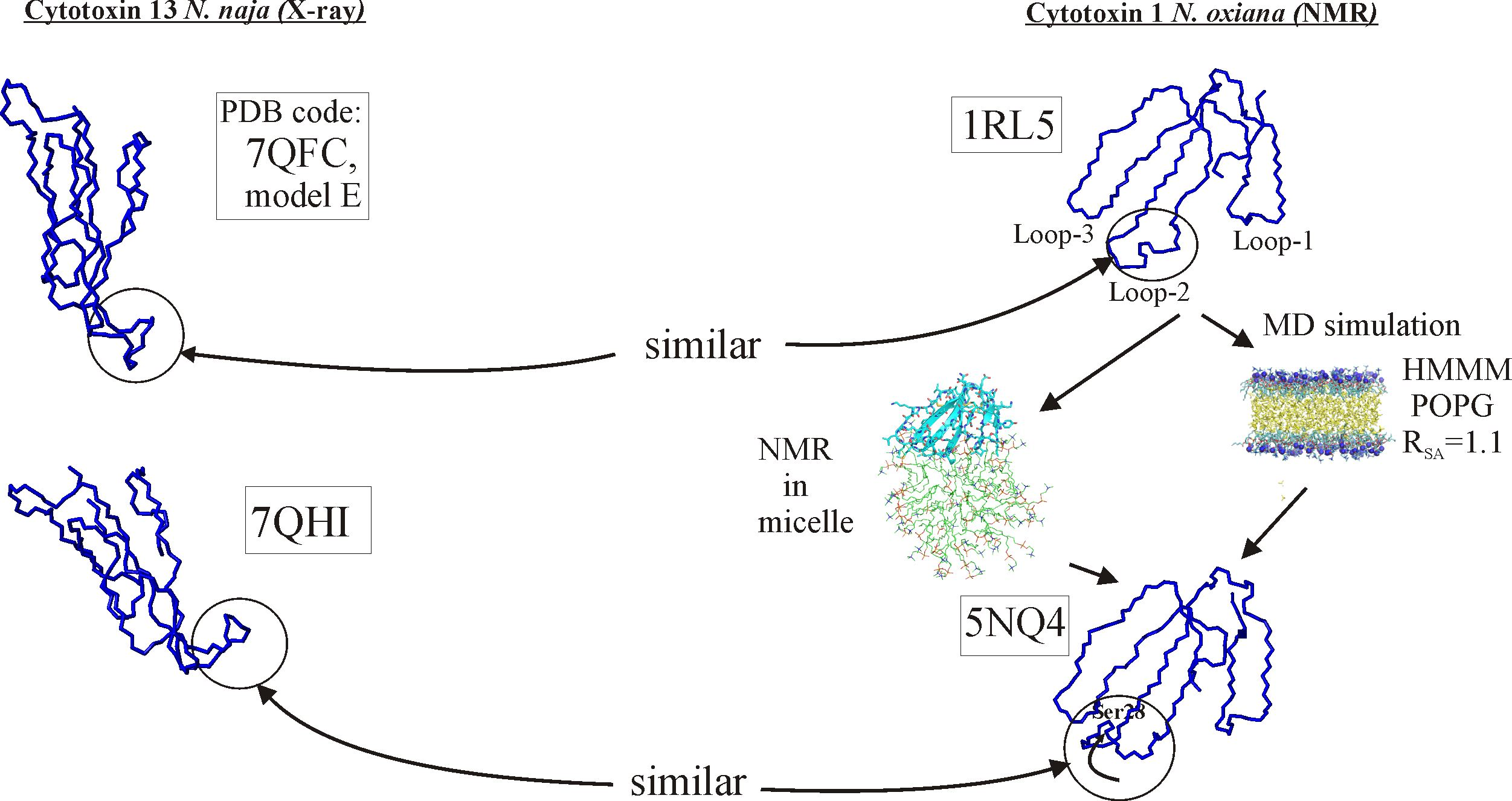
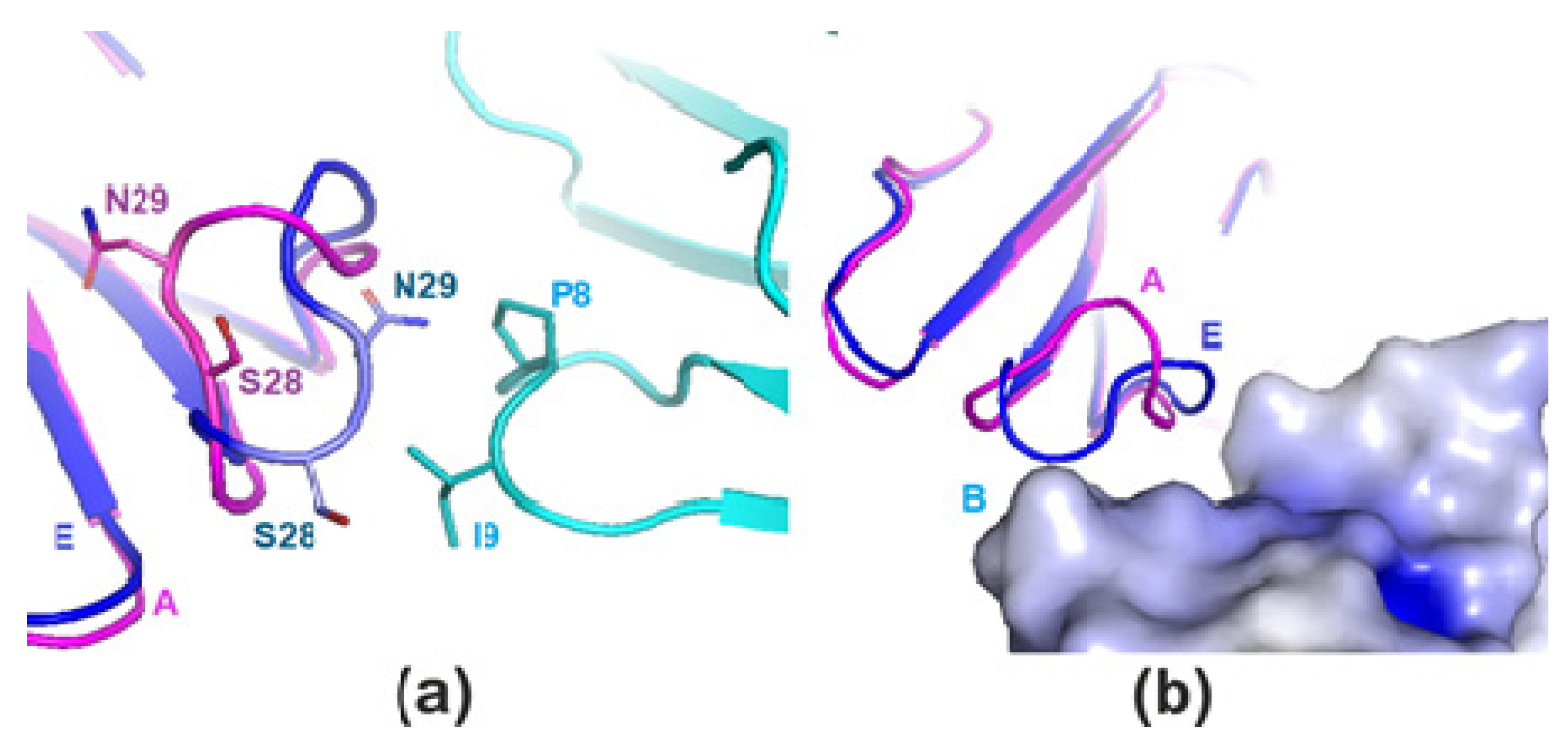
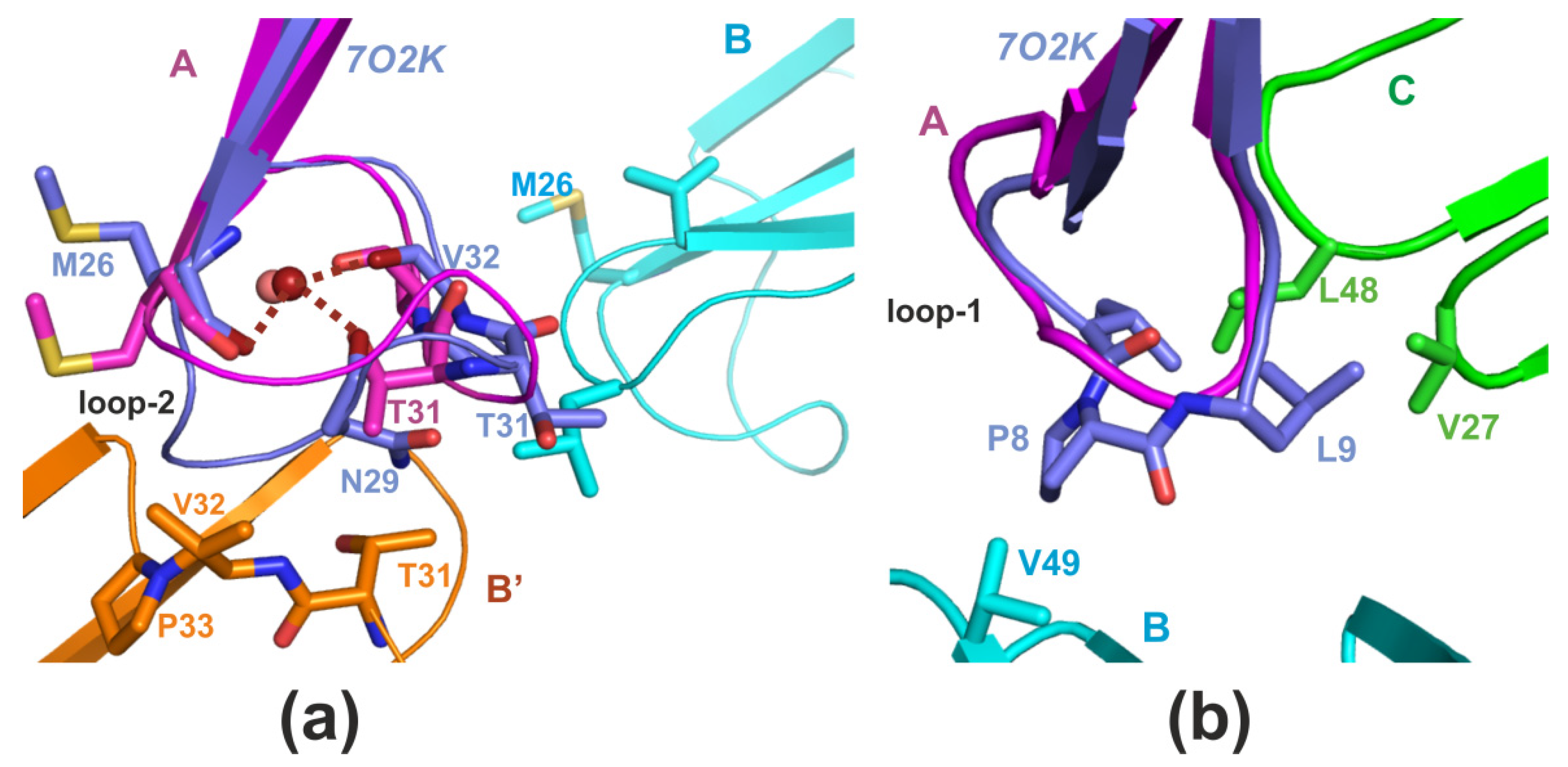
2.1. X-ray Crystallography
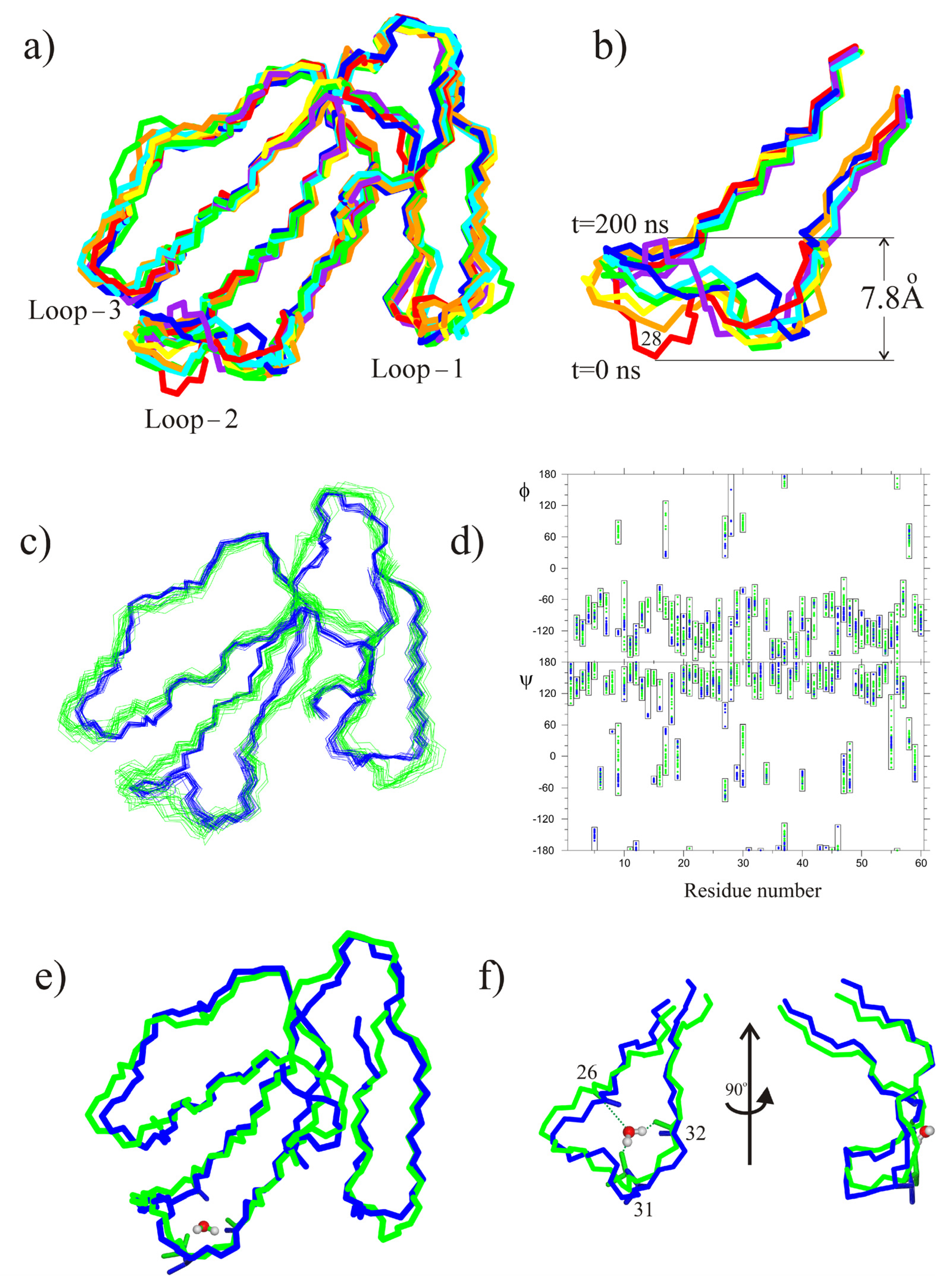
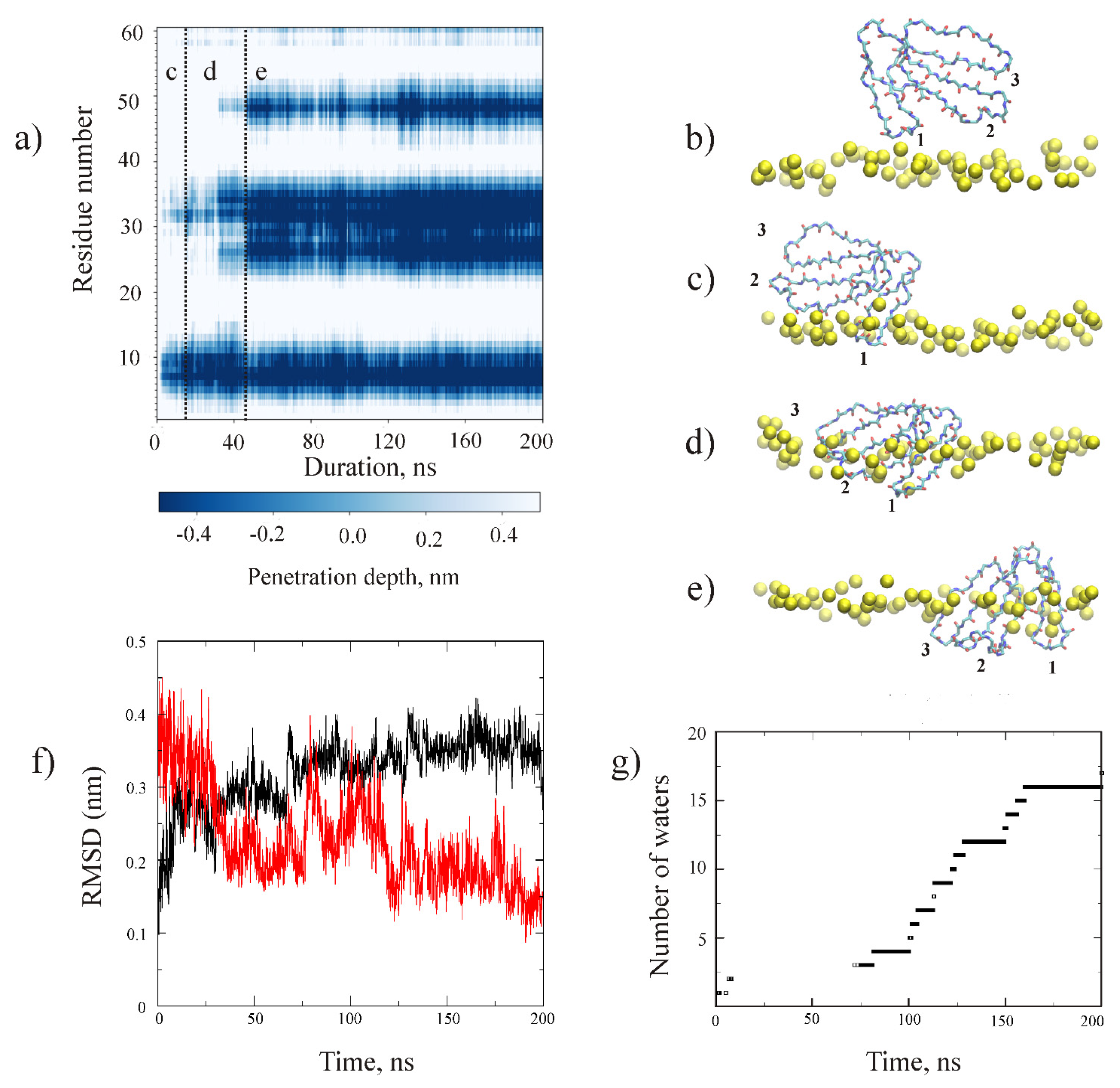
2.2. MD Study of CT13Nn and CT1No/HMMM POPG Interactions
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cytotoxin Purification
5.2. Crystallization, Data Collection, and Processing
5.3. Structure Determination and Refinement
5.4. Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wong, K.Y.; Tan, C.H.; Tan, K.Y.; Quraishi, N.H.; Tan, N.H. Elucidating the biogeographical variation of the venom of Naja naja (spectacled cobra) from Pakistan through a venom-decomplexing proteomic study. J. Proteom. 2018, 175, 156–173. [Google Scholar] [CrossRef]
- Dutta, S.; Chanda, A.; Kalita, B.; Islam, T.; Patra, A.; Mukherjee, A.K. Proteomic analysis to unravel the complex venom proteome of eastern India Naja naja: Correlation of venom composition with its biochemical and pharmacological properties. J. Proteom. 2017, 156, 29–39. [Google Scholar] [CrossRef]
- Petras, D.; Sanz, L.; Segura, A.; Herrera, M.; Villalta, M.; Solano, D.; Vargas, M.; Leon, G.; Warrell, D.A.; Theakston, R.D.G.; et al. Snake venomics of African spitting cobras: Toxin composition and assessment of congeneric cross-reactivity of the Pan-African EchiTAb-Plus-ICP antivenom by antivenomics and neutralization approaches. J. Proteome Res. 2011, 10, 1266–1280. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, C.H.; Fung, S.Y.; Tan, N.H. Venomics, lethality and neutralization of Naja kaouthia (monocled cobra) venoms from three different geographical regions of Southeast Asia. J. Proteom. 2015, 120, 105–125. [Google Scholar] [CrossRef]
- Ziganshin, R.H.; Kovalchuk, S.I.; Arapidi, G.P.; Starkov, V.G.; Hoang, A.N.; Nguyen, T.T.T.; Nguyen, K.C.; Shoibonov, B.B.; Tsetlin, V.I.; Utkin, Y.N. Quantitative proteomic analysis of Vietnamese krait venoms: Neurotoxins are the major components in Bungarus multicinctus and phospholipases A2 in Bungarus fasciatus. Toxicon 2015, 107, 197–209. [Google Scholar] [CrossRef]
- Tan, C.H.; Fung, S.Y.; Yap, M.K.K.; Leong, P.K.; Liew, J.L.; Tan, N.H. Unveiling the elusive and exotic: Venomics of the Malayan blue coral snake (Calliophis bivirgata flaviceps). J. Proteom. 2016, 132, 1–12. [Google Scholar] [CrossRef]
- Konshina, A.G.; Dubovskii, P.V.; Efremov, R.G. Structure and dynamics of cardiotoxins. Curr. Protein Pept. Sci. 2012, 13, 570–584. [Google Scholar] [CrossRef]
- Galat, A.; Gross, G.; Drevet, P.; Sato, A.; Menez, A. Conserved structural determinants in three-fingered protein domains. FEBS J. 2008, 275, 3207–3225. [Google Scholar] [CrossRef]
- Kini, R.M. Molecular moulds with multiple missions: Functional sites in three-finger toxins. Clin. Exp. Pharmcol. Physiol. 2002, 29, 815–822. [Google Scholar] [CrossRef]
- Kumar, T.K.; Jayaraman, G.; Lee, C.S.; Arunkumar, A.I.; Sivaraman, T.; Samuel, D.; Yu, C. Snake venom cardiotoxins-structure, dynamics, function and folding. J. Biomol. Struct. Dyn. 1997, 15, 431–463. [Google Scholar] [CrossRef]
- Tsetlin, V.I.; Kasheverov, I.E.; Utkin, Y.N. Three-finger proteins from snakes and humans acting on nicotinic receptors: Old and new. J. Neurochem. 2020, 158, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Nirthanan, S. Snake three-finger α-neurotoxins and nicotinic acetylcholine receptors: Molecules, mechanisms and medicine. Biochem. Pharmcol. 2020, 181, 114168. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M.; Koh, C.Y. Snake venom three-finger toxins and their potential in drug development targeting cardiovascular diseases. Biochem. Pharmcol. 2020, 181, 114105. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Xu, J.; Kadji, F.M.N.; Clark, M.J.; Zhao, J.; Tsutsumi, N.; Aoki, J.; Sunahara, R.K.; Inoue, A.; Garcia, K.C.; et al. Structure and selectivity engineering of the M1 muscarinic receptor toxin complex. Science 2020, 369, 161–167. [Google Scholar] [CrossRef]
- Kini, R.M. Toxins for decoding interface selectivity in nicotinic acetylcholine receptors. Biochem. J. 2019, 476, 1515–1520. [Google Scholar] [CrossRef]
- Utkin, Y.N. Last decade update for three-finger toxins: Newly emerging structures and biological activities. World J. Biol. Chem. 2019, 10, 17–27. [Google Scholar] [CrossRef]
- Dubovskii, P.V.; Efremov, R.G. The role of hydrophobic /hydrophilic balance in the activity of structurally flexible vs. rigid cytolytic polypeptides and analogs developed on their basis. Expert Rev. Proteom. 2018, 15, 873–886. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Dagda, R.K.; Rael, E.D. Snake venom cytotoxins, phospholipase A2s, and Zn-dependent metalloproteinases: Mechanisms of action and pharmacological relevance. J. Clin. Toxicol. 2014, 4, 1000181. [Google Scholar] [CrossRef] [Green Version]
- Dubovskii, P.V.; Utkin, Y.N. Antiproliferative activity of cobra venom cytotoxins. Curr. Top. Med. Chem. 2015, 15, 638–648. [Google Scholar] [CrossRef]
- Chien, K.Y.; Chiang, C.M.; Hseu, Y.C.; Vyas, A.A.; Rule, G.S.; Wu, W. Two distinct types of cardiotoxin as revealed by the structure and activity relationship of their interaction with zwitterionic phospholipid dispersions. J. Biol. Chem. 1994, 269, 14473–14483. [Google Scholar] [CrossRef]
- Dubovskii, P.V.; Dementieva, D.V.; Bocharov, E.V.; Utkin, Y.N.; Arseniev, A.S. Membrane binding motif of the P-type cardiotoxin. J. Mol. Biol. 2001, 305, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Dubovskii, P.V.; Dubinny, M.A.; Volynsky, P.E.; Pustovalova, Y.E.; Konshina, A.G.; Utkin, Y.N.; Efremov, R.G.; Arseniev, A.S. Impact of membrane partitioning on the spatial structure of an S-type cobra cytotoxin. J. Biomol. Struct. Dyn. 2018, 36, 3463–3478. [Google Scholar] [CrossRef] [PubMed]
- Dauplais, M.; Neumann, J.M.; Pinkasfeld, S.; Menez, A.; Roumestand, C. An NMR study of the interaction of cardiotoxin gamma from Naja nigricollis with perdeuterated dodecylphosphocholine micelles. Eur. J. Biochem. 1995, 230, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gorai, B.; Karthikeyan, M.; Sivaraman, T. Putative membrane lytic sites of P-type and S-type cardiotoxins from snake venoms as probed by all-atom molecular dynamics simulations. J. Mol. Model. 2016, 22, 238. [Google Scholar] [CrossRef]
- Dubinnyi, M.A.; Lesovoy, D.M.; Dubovskii, P.V.; Chupin, V.V.; Arseniev, A.S. Modeling of 31P-NMR spectra of magnetically oriented phospholipid liposomes: A new analytical solution. Solid State Nucl. Magn. Reson. 2006, 29, 305–311. [Google Scholar] [CrossRef]
- Dubovskii, P.V.; Lesovoy, D.M.; Dubinnyi, M.A.; Utkin, Y.N.; Arseniev, A.S. Interaction of the P-type cardiotoxin with phospholipid membranes. Eur. J. Biochem. 2003, 270, 2038–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubovskii, P.V.; Lesovoy, D.M.; Dubinnyi, M.A.; Konshina, A.G.; Utkin, Y.N.; Efremov, R.G.; Arseniev, A.S. Interaction of three-finger toxins with phospholipid membranes: Comparison of S- and P-type cytotoxins. Biochem. J. 2005, 387, 807–815. [Google Scholar] [CrossRef]
- Su, Z.-Y.; Wang, Y.-T. Coarse-Grained Molecular Dynamics simulations of cobra cytotoxin A3 interactions with a lipid bilayer: Penetration of loops into membranes. J. Phys. Chem. B 2011, 115, 796–802. [Google Scholar] [CrossRef]
- Levtsova, O.V.; Antonov, M.Y.; Mordvintsev, D.Y.; Utkin, Y.N.; Shaitan, K.V.; Kirpichnikov, M.P. Steered molecular dynamics simulations of cobra cytotoxin interaction with zwitterionic lipid bilayer: No penetration of loop tips into membranes. Comput. Biol. Chem. 2009, 33, 29–32. [Google Scholar] [CrossRef]
- Gorai, B.; Sivaraman, T. Delineating residues for haemolytic activities of snake venom cardiotoxin 1 from Naja naja as probed by molecular dynamics simulations and in vitro validations. Int. J. Biol. Macromol. 2017, 95, 1022–1036. [Google Scholar] [CrossRef]
- Konshina, A.G.; Dubovskii, P.V.; Efremov, R.G. Stepwise insertion of cobra cardiotoxin CT2 into a lipid bilayer occurs as an interplay of protein and membrane “Dynamic Molecular Portraits”. J. Chem. Inf. Model. 2021, 61, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Shulepko, M.A.; Lyukmanova, E.N.; Shenkarev, Z.O.; Dubovskii, P.V.; Astapova, M.V.; Feofanov, A.V.; Arseniev, A.S.; Utkin, Y.N.; Kirpichnikov, M.P.; Dolgikh, D.A. Towards universal approach for bacterial production of three-finger Ly6/uPAR proteins: Case study of cytotoxin I from cobra N. oxiana. Protein Expr. Purif. 2017, 130, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Chen, S.T.; Ho, C.L.; Wang, K.T. Synthesis of a fully active snake-venom cardiotoxin by fragment condensation on solid polymer. Biochim. Biophys. Acta 1978, 536, 376–389. [Google Scholar] [CrossRef]
- Huang, S.Y.; Wei, T.Y.; Liu, B.S.; Lin, M.H.; Chiang, S.K.; Chen, S.F.; Sung, W.C. Monitoring the disulfide bonds of folding isomers of synthetic CTX A3 polypeptide using MS-based technology. Toxins 2019, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Silva-de-Franca, F.; Villas-Boas, I.M.; Serrano, S.M.D.; Cogliati, B.; Chudzinski, S.A.D.; Lopes, P.H.; Kitano, E.S.; Okamoto, C.K.; Tambourgi, D.V. Naja annulifera snake: New insights into the venom components and pathogenesis of envenomation. PLoS Negl. Trop. Dis. 2019, 13, e0007017. [Google Scholar] [CrossRef] [Green Version]
- Dubovskii, P.V.; Ignatova, A.A.; Feofanov, A.V.; Utkin, Y.N.; Efremov, R.G. Antibacterial activity of cardiotoxin-like basic polypeptide from cobra venom. Bioorg. Med. Chem. Lett. 2020, 30, 126890. [Google Scholar] [CrossRef]
- Dubinnyi, M.A.; Dubovskii, P.V.; Starkov, V.A.; Utkin, Y.N. The omega-loop of cobra cytotoxins tolerates multiple amino acid substitutions. Biochem. Biophys. Res. Commun. 2021, 558, 141–146. [Google Scholar] [CrossRef]
- Ohkubo, Y.Z.; Pogorelov, T.V.; Arcario, M.J.; Christensen, G.A.; Tajkhorshid, E. Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J. 2012, 102, 2130–2139. [Google Scholar] [CrossRef] [Green Version]
- Dubovskii, P.V.; Dubinnyi, M.A.; Konshina, A.G.; Kazakova, E.D.; Sorokoumova, G.M.; Ilyasova, T.M.; Shulepko, M.A.; Chertkova, R.V.; Lyukmanova, E.N.; Dolgikh, D.A.; et al. Structural and dynamic “Portraits” of recombinant and native cytotoxin I from Naja oxiana: How close are they? Biochemistry 2017, 56, 4468–4477. [Google Scholar] [CrossRef]
- Dementieva, D.V.; Bocharov, E.V.; Arseniev, A.S. Two forms of cytotoxin II (cardiotoxin) from Naja naja oxiana in aqueous solution: Spatial structures with tightly bound water molecules. Eur. J. Biochem. 1999, 263, 152–162. [Google Scholar] [CrossRef]
- Lee, C.S.; Kumar, T.K.S.; Lian, L.Y.; Cheng, J.W.; Yu, C. Main-chain dynamics of cardiotoxin II from Taiwan cobra (Naja naja atra) as studied by carbon-13 NMR at natural abundance: Delineation of the role of functionally important residues. Biochemistry 1998, 37, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Dubinnyi, M.A.; Dubovskii, P.V.; Utkin, Y.N.; Simonova, T.N.; Barsukov, L.I.; Arseniev, A.S. An ESR study of the cytotoxin II interaction with model membranes. Russ. J. Bioorg. Chem. 2001, 27, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Leszczynski, J.F.; Rose, G.D. Loops in globular proteins: A novel category of secondary structure. Science 1986, 234, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Fetrow, J.S. Omega loops: Nonregular secondary structures significant in protein function and stability. FASEB J. 1995, 9, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Wu, W.G.; Chiang, C.M.; Hsin, A.Y.; Hsiao, C.D. Crystal structure of cardiotoxin V from Taiwan cobra venom: pH-dependent conformational change and a novel membrane-binding motif identified in the three-finger loops of P-type cardiotoxin. Biochemistry 1997, 36, 2403–2413. [Google Scholar] [CrossRef]
- Gillespie, C.M.; Asthagiri, D.; Lenhoff, A.M. Polymorphic protein crystal growth: Influence of hydration and ions in glucose isomerase. Cryst. Growth Des. 2014, 14, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Dubovskii, P.V.; Konshina, A.G.; Efremov, R.G. Cobra cardiotoxins: Membrane interactions and pharmacological potential. Curr. Med. Chem. 2014, 21, 270–287. [Google Scholar] [CrossRef]
- Sandoval-Perez, A.; Pluhackova, K.; Bockmann, R.A. Critical comparison of biomembrane force fields: Protein-lipid interactions at the membrane interface. J. Chem. Theory Comput. 2017, 13, 2310–2321. [Google Scholar] [CrossRef]
- Feofanov, A.V.; Sharonov, G.V.; Dubinnyi, M.A.; Astapova, M.V.; Kudelina, I.A.; Dubovskii, P.V.; Rodionov, D.I.; Utkin, Y.N.; Arseniev, A.S. Comparative study of structure and activity of cytotoxins from venom of the cobras Naja oxiana, Naja kaouthia, and Naja haje. Biochemistry 2004, 69, 1148–1157. [Google Scholar] [CrossRef]
- Chong, H.P.; Tan, K.Y.; Tan, C.H. Cytotoxicity of snake venoms and cytotoxins from two Southeast Asian cobras (Naja sumatrana, Naja kaouthia): Exploration of anticancer potential, selectivity, and cell death mechanism. Front Mol. Biosci. 2020, 7, 583587. [Google Scholar] [CrossRef]
- Averin, A.S.; Nenov, M.N.; Starkov, V.G.; Tsetlin, V.I.; Utkin, Y.N. Effects of cardiotoxins from Naja oxiana cobra venom on rat heart muscle and aorta: A comparative study of toxin-induced contraction mechanisms. Toxins 2022, 14, 88. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Tatematsu, Y.; Tabata, A.; Nagamune, H.; Ohkura, K. Cytolytic activity and molecular feature of cardiotoxin and cardiotoxin-like basic protein: The Electrostatic potential field is an important factor for cell lytic activity. Anticancer Res. 2015, 35, 4515–4519. [Google Scholar] [PubMed]
- Kuleshina, O.N.; Kruykova, E.V.; Cheremnykh, E.G.; Kozlov, L.V.; Andreeva, T.V.; Starkov, V.G.; Osipov, A.V.; Ziganshin, R.H.; Tsetlin, V.I.; Utkin, Y.N. Screening snake venoms for toxicity to Tetrahymena Pyriformis revealed anti-protozoan activity of cobra cytotoxins. Toxins 2020, 12, 325. [Google Scholar] [CrossRef]
- Utkin, Y.N.; Kuch, U.; Kasheverov, I.E.; Lebedev, D.S.; Cederlund, E.; Molles, B.E.; Polyak, I.; Ivanov, I.A.; Prokopev, N.A.; Ziganshin, R.H.; et al. Novel long-chain neurotoxins from Bungarus candidus distinguish the two binding sites in muscle-type nicotinic acetylcholine receptors. Biochem. J. 2019, 476, 1285–1302. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Cryst. 2010, D66, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otwinowski, Z.; Minor, W.I. Methods in Enzymology; Academic Press: New York, NY, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997, D53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. 2010, D66, 486–501. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Qi, Y.; Cheng, X.; Lee, J.; Vermaas, J.V.; Pogorelov, T.V.; Tajkhorshid, E.; Park, S.; Klauda, J.B.; Im, W. CHARMM-GUI HMMM builder for membrane simulations with the Highly Mobile Membrane-Mimetic Model. Biophys. J. 2015, 109, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Dubovskii, P.V.; Ignatova, A.A.; Volynsky, P.E.; Ivanov, I.A.; Zhmak, M.N.; Feofanov, A.V.; Efremov, R.G. Improving therapeutic potential of antibacterial spider venom peptides: Coarse-grain molecular dynamics guided approach. Future Med. Chem. 2018, 10, 2309–2322. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Name/UNIPROT Code | Amino Acid Sequence (with Numbering above the Sequence) |
|---|---|
| 1 5 10 15 20 25 30 35 40 45 50 55 60 | |
| CT3Nk 1/P01446 | LKCNKLIPLAYKTCPAGKNLCYKMFMVSNKTVPVKRGCIDACPKNSLLVKYVCCNTDRCN |
| CT13Nn 2 | LKCNKLIPLAYKTCPAGKNLCYKMFMVSNKTVPVKRGCIDVCPKNSLLVKYVCCNTDRCN |
| CT1No 3/P01451 | LKCNKLVPIAYKTCPEGKNLCYKMFMMSDLTIPVKRGCIDVCPKNSLLVKYVCCNTDRCN |
| CT3Nn/P24780 | LKCNKLIPLAYKTCPAGKNLCYKMFMVSNKTVPVKRGCIDVCPKNSLVLKYVCCNTDRCN |
| Crystal Form | Hexagonal | Orthorhombic |
|---|---|---|
| Data collection | ||
| Synchrotron | SPring-8 | DESY, Petra III |
| Wavelength (Å) | 1.000 | 0.9763 |
| Space group | P6422 | C2221 |
| a, b, c (Å) | 109.33, 109.33, 135.77 | 88.74, 97.17, 132.12 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 |
| Resolution range (Å) | 50.0–2.30 (2.36–2.30) * | 50.0–2.50 (2.63–2.60) * |
| Rmege (%) | 10.4 (54.7) | 18.1 (89.4) |
| <I/σ(I)> | 13.4 (2.3) | 10.7 (1.8) |
| Completeness (%) | 98.39 (89.48) | 99.17 (99.93) |
| Redundancy | 8.2 (7.2) | 6.3 (3.4) |
| Refinement | ||
| Resolution (Å) | 14.95–2.30 (2.36–2.30) * | 14.91–2.60 (2.66–2.60) * |
| Number of reflections | 20,428 | 18,995 |
| Rwork/Rfree (%) | 19.7/24.2 | 20.5/25.5 |
| No. atoms | ||
| Protein | 1392 | 2784 |
| Water | 218 | 17 |
| B-factors | ||
| Protein | 38.11 | 98.51 |
| Solvent | 35.98 | 43.93 |
| RMSD | ||
| Bond lengths (Å) | 0.013 | 0.013 |
| Bond angles (°) | 2.094 | 2.191 |
| Ramachandran statistics | ||
| Most-favored regions (%) | 94.3 | 89.4 |
| Allowed regions (%) | 4.0 | 8.3 |
| Disallowed regions (%) | 1.7 | 2.3 |
| Toxin | |||
|---|---|---|---|
| CT1No a | CT13Nn a | ||
| Solution b | Membrane c | Solution b | Membrane |
| 1RL5 | 5NQ4 | 7O2K | 7QHI (model A) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubovskii, P.V.; Dubova, K.M.; Bourenkov, G.; Starkov, V.G.; Konshina, A.G.; Efremov, R.G.; Utkin, Y.N.; Samygina, V.R. Variability in the Spatial Structure of the Central Loop in Cobra Cytotoxins Revealed by X-ray Analysis and Molecular Modeling. Toxins 2022, 14, 149. https://doi.org/10.3390/toxins14020149
Dubovskii PV, Dubova KM, Bourenkov G, Starkov VG, Konshina AG, Efremov RG, Utkin YN, Samygina VR. Variability in the Spatial Structure of the Central Loop in Cobra Cytotoxins Revealed by X-ray Analysis and Molecular Modeling. Toxins. 2022; 14(2):149. https://doi.org/10.3390/toxins14020149
Chicago/Turabian StyleDubovskii, Peter V., Kira M. Dubova, Gleb Bourenkov, Vladislav G. Starkov, Anastasia G. Konshina, Roman G. Efremov, Yuri N. Utkin, and Valeriya R. Samygina. 2022. "Variability in the Spatial Structure of the Central Loop in Cobra Cytotoxins Revealed by X-ray Analysis and Molecular Modeling" Toxins 14, no. 2: 149. https://doi.org/10.3390/toxins14020149
APA StyleDubovskii, P. V., Dubova, K. M., Bourenkov, G., Starkov, V. G., Konshina, A. G., Efremov, R. G., Utkin, Y. N., & Samygina, V. R. (2022). Variability in the Spatial Structure of the Central Loop in Cobra Cytotoxins Revealed by X-ray Analysis and Molecular Modeling. Toxins, 14(2), 149. https://doi.org/10.3390/toxins14020149