Aryl Hydrocarbon Receptor Inhibition Restores Indoxyl Sulfate-Mediated Endothelial Dysfunction in Rat Aortic Rings


Abstract
:1. Introduction
2. Results
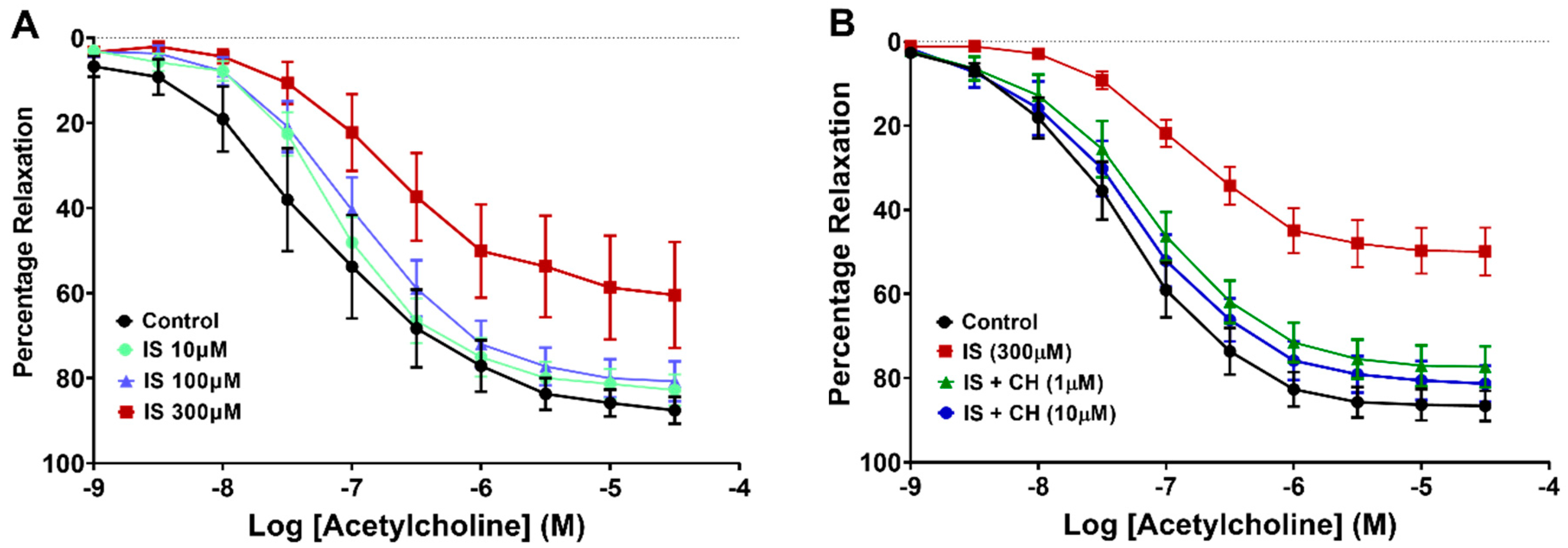
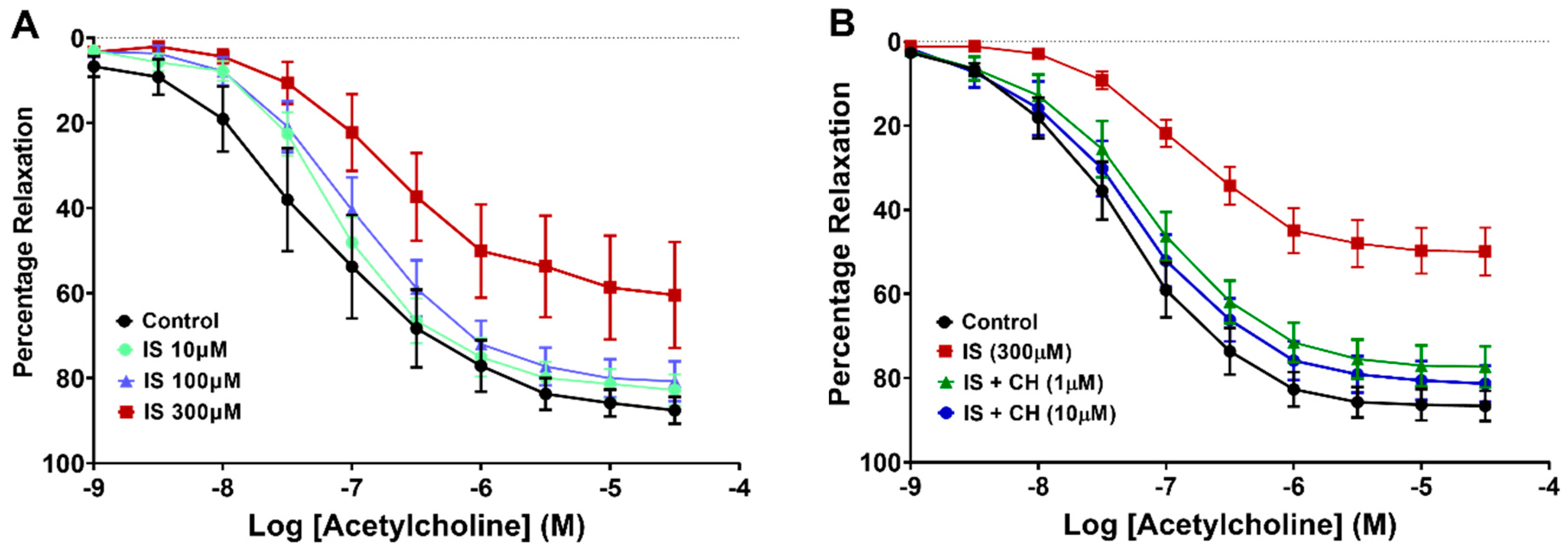
2.1. Effect of Aryl Hydrocarbon Receptor Inhibition on IS-Impaired Endothelium-Dependent Relaxation
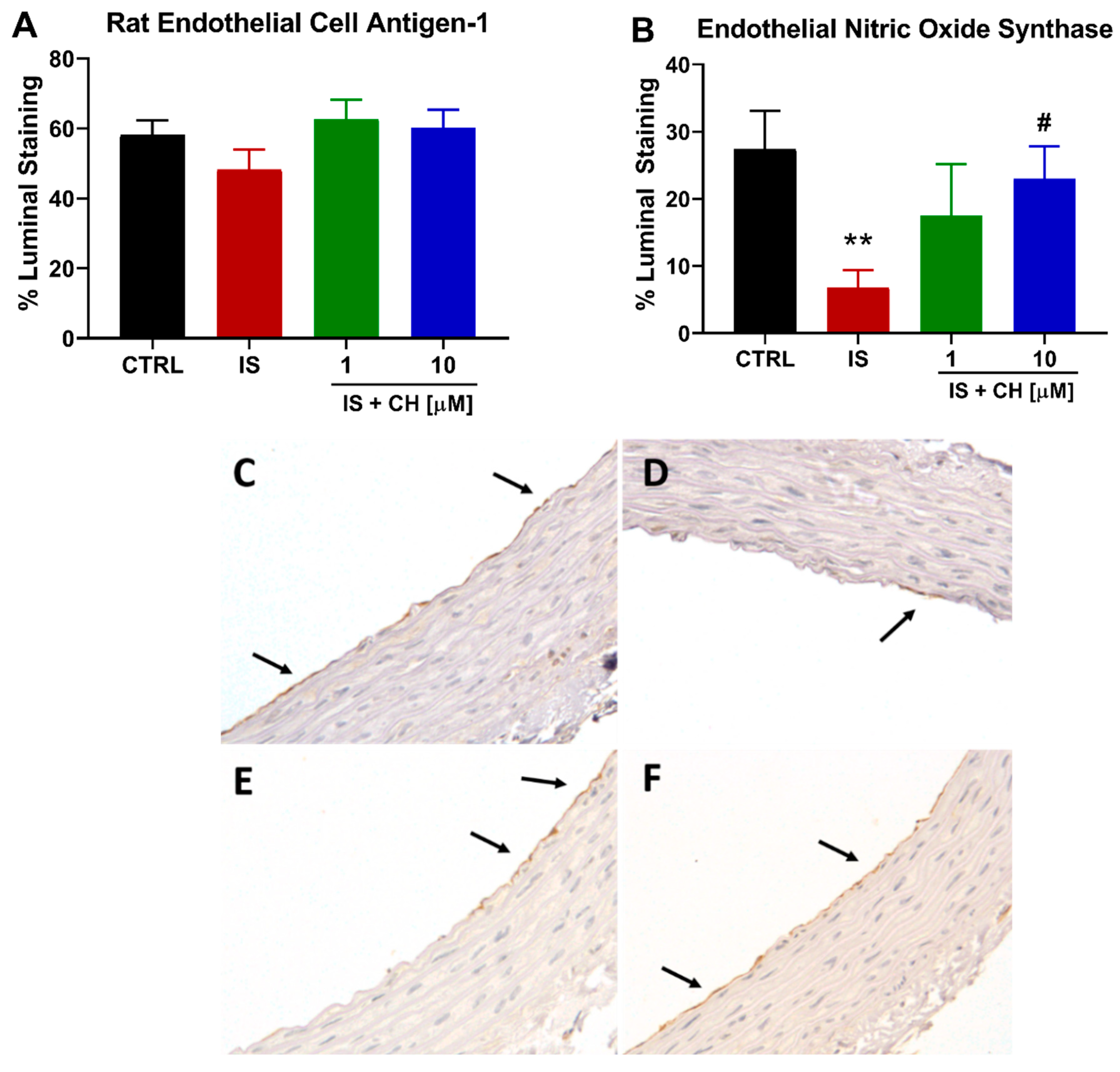
2.2. Effect of IS on the Endothelium and Endothelial Nitric Oxide Synthase
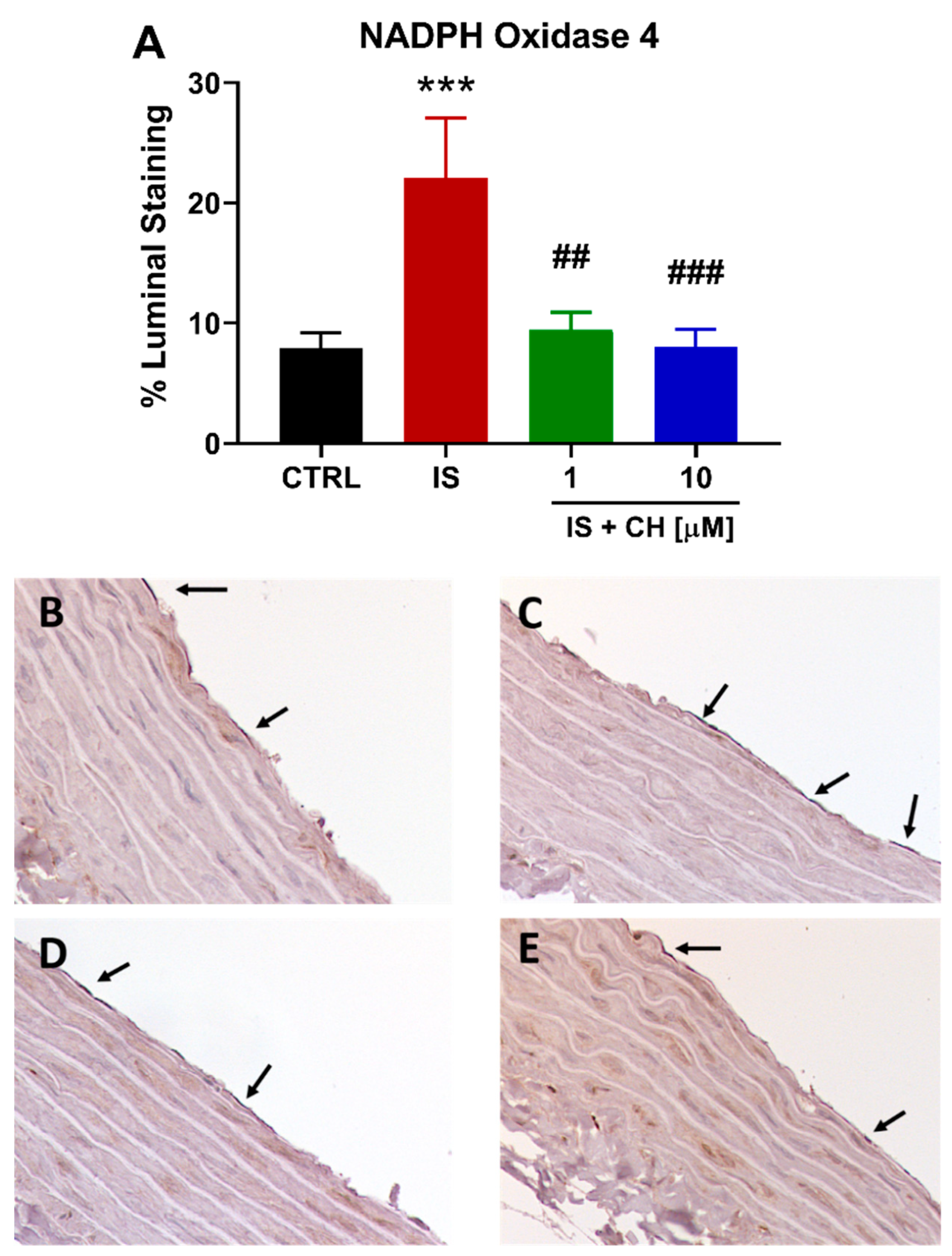
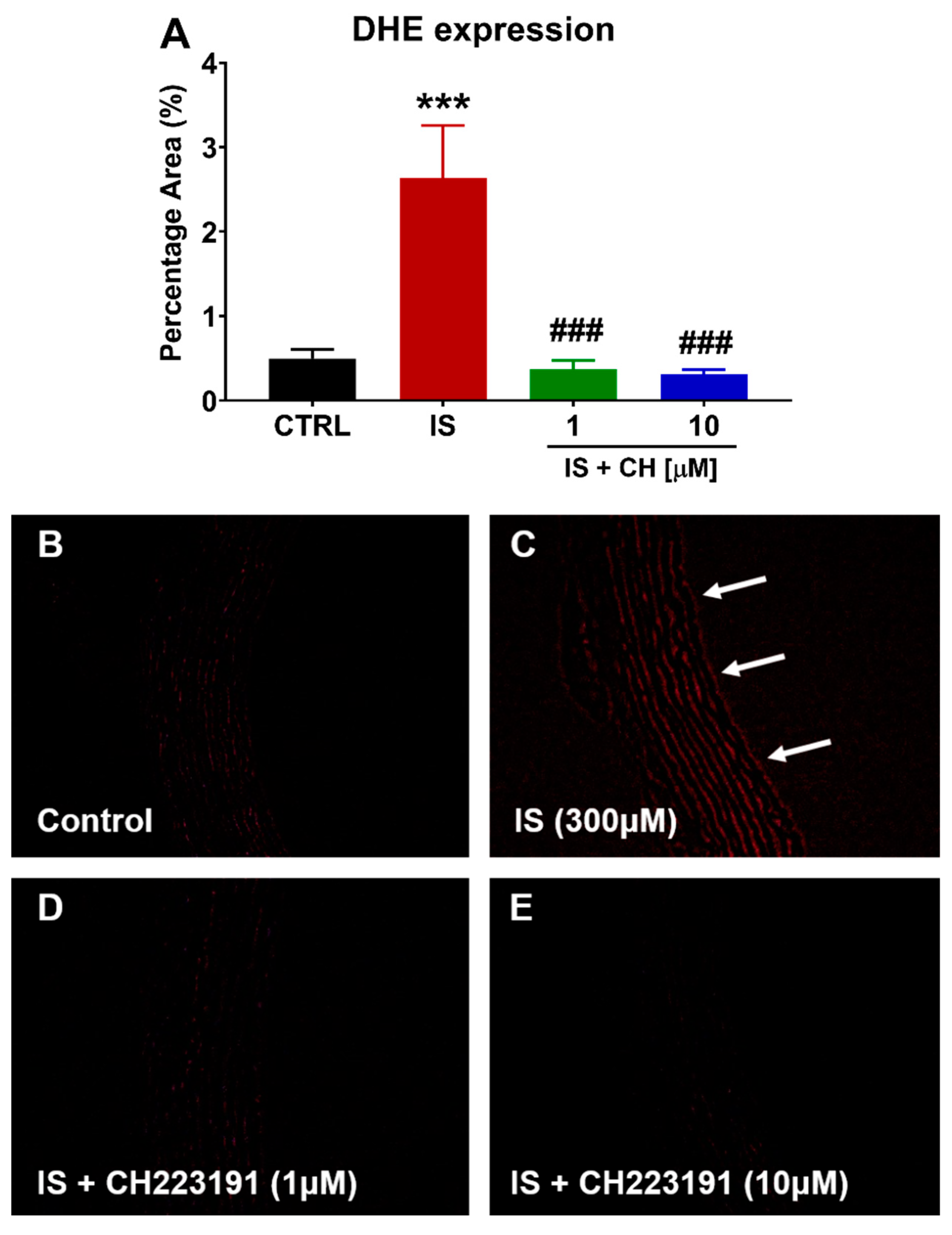
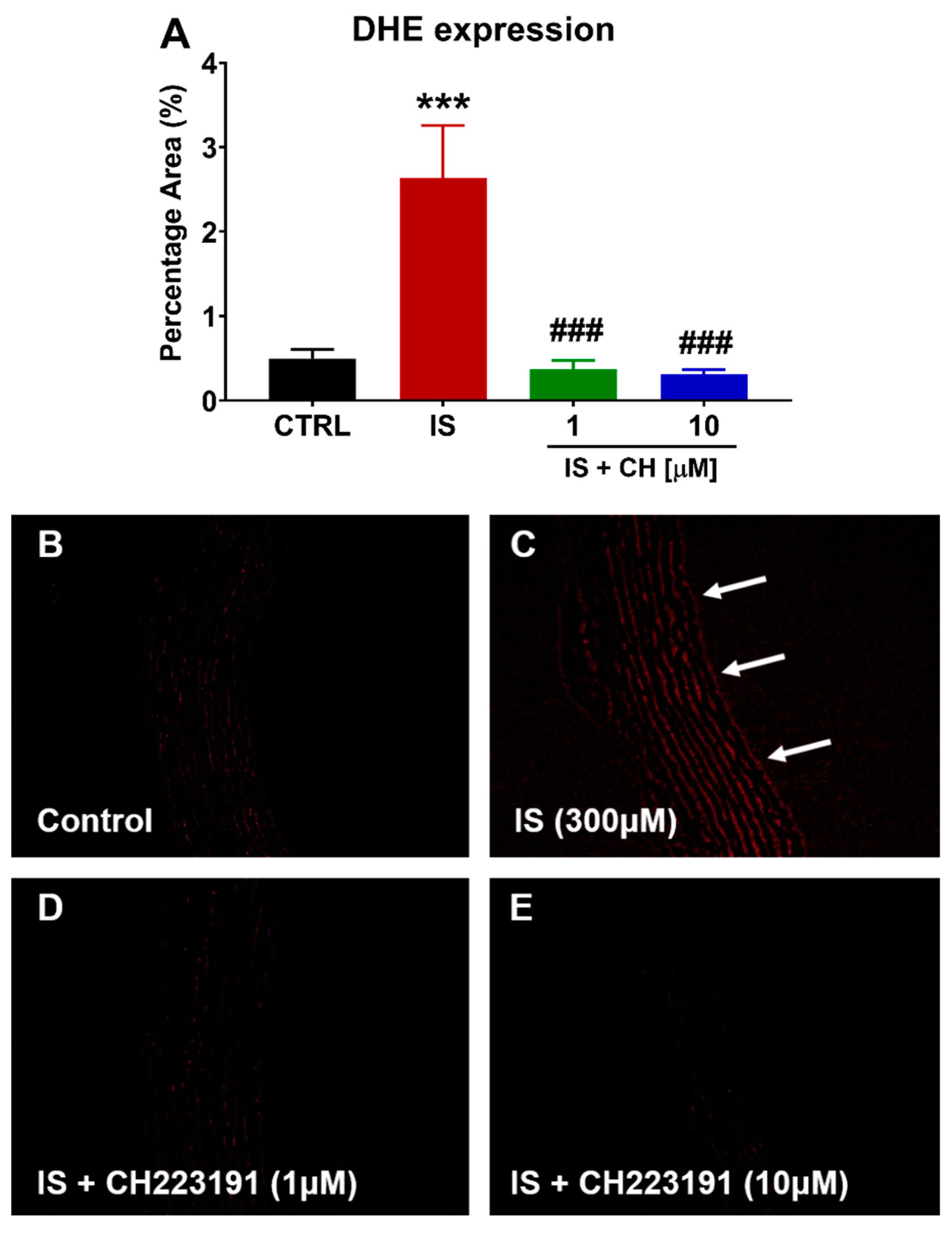
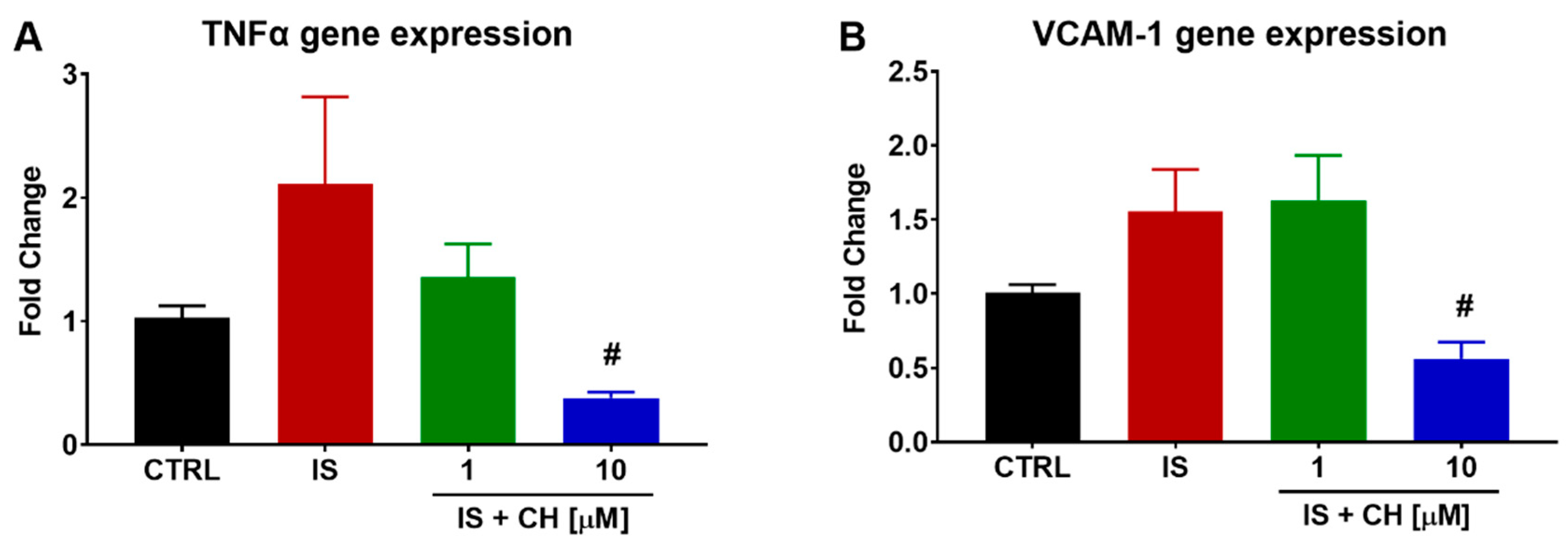
2.3. Effect of IS and CH223191 on Pro- and Anti-Oxidative Gene Expression
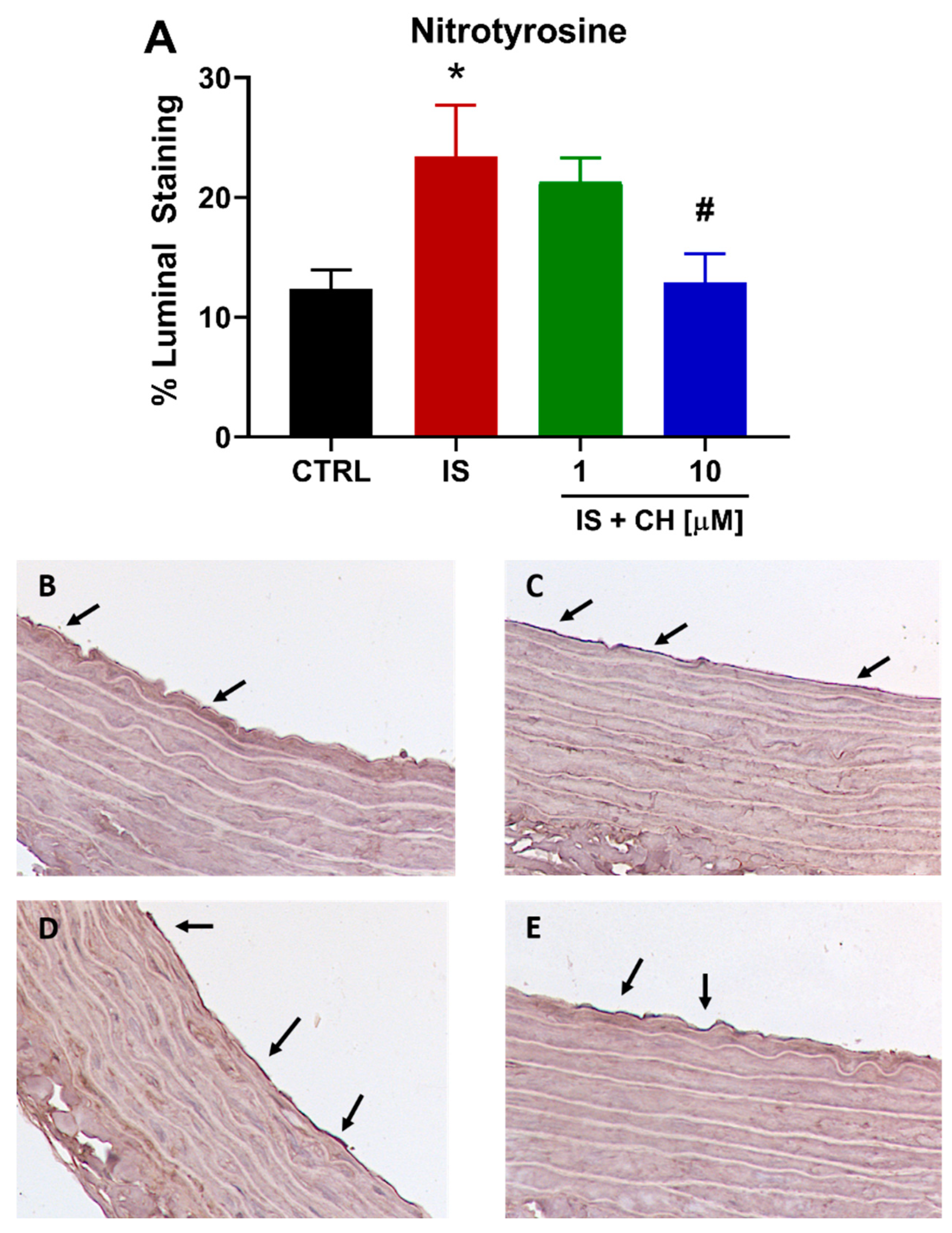
2.4. Effect of IS and CH223191 on Markers of Oxidative Stress and Inflammation
3. Discussion
4. Materials and Methods
4.1. Vascular Reactivity
4.2. Gene Expression Studies
4.3. Immunohistochemistry
4.4. Assessment of Reactive Oxygen Species
4.5. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savira, F.; Ademi, Z.; Wang, B.H.; Kompa, A.R.; Owen, A.J.; Liew, D.; Zomer, E. The Preventable Productivity Burden of Kidney Disease in Australia. J. Am. Soc. Nephrol. 2021, 32, 938–949. [Google Scholar] [CrossRef]
- Elshahat, S.; Cockwell, P.; Maxwell, A.P.; Griffin, M.; O’Brien, T.; O’Neill, C. The impact of chronic kidney disease on developed countries from a health economics perspective: A systematic scoping review. PLoS ONE 2020, 15, e0230512. [Google Scholar] [CrossRef] [Green Version]
- Manjunath, G.; Tighiouart, H.; Ibrahim, H.; MacLeod, B.; Salem, D.N.; Griffith, J.L.; Coresh, J.; Levey, A.S.; Sarnak, M.J. Level of kidney function as a risk factor for atherosclerotic cardiovascular outcomes in the community. J. Am. Coll. Cardiol. 2003, 41, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imazu, M.; Fukuda, H.; Kanzaki, H.; Amaki, M.; Hasegawa, T.; Takahama, H.; Hitsumoto, T.; Tsukamoto, O.; Morita, T.; Ito, S.; et al. Plasma indoxyl sulfate levels predict cardiovascular events in patients with mild chronic heart failure. Sci. Rep. 2020, 10, 16528. [Google Scholar] [CrossRef]
- Di Lullo, L.; House, A.; Gorini, A.; Santoboni, A.; Russo, D.; Ronco, C. Chronic kidney disease and cardiovascular complications. Heart Fail. Rev. 2015, 20, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular incompetence in dialysis patients--protein-bound uremic toxins and endothelial dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, Q.L.; Tow, F.K.; Deva, R.; Alias, M.A.; Kawasaki, R.; Wong, T.Y.; Mohamad, N.; Colville, D.; Hutchinson, A.; Savige, J. The microvasculature in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 1872–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl sulfate potentiates endothelial dysfunction via reciprocal role for reactive oxygen species and RhoA/ROCK signaling in 5/6 nephrectomized rats. Free Radic. Res. 2017, 51, 237–252. [Google Scholar] [CrossRef]
- Six, I.; Gross, P.; Remond, M.C.; Chillon, J.M.; Poirot, S.; Drueke, T.B.; Massy, Z.A. Deleterious vascular effects of indoxyl sulfate and reversal by oral adsorbent AST-120. Atherosclerosis 2015, 243, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Kompa, A.R.; Wang, B.H.; Kelly, D.J.; Krum, H. Cardiorenal syndrome: The emerging role of protein-bound uremic toxins. Circ. Res. 2012, 111, 1470–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernomian, L.; da Silva, C. Current basis for discovery and development of aryl hydrocarbon receptor antagonists for experimental and therapeutic use in atherosclerosis. Eur. J. Pharmacol. 2015, 764, 118–123. [Google Scholar] [CrossRef]
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of aryl hydrocarbon receptor mediates indoxyl sulfate-induced monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells. Circ. J. 2013, 77, 224–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Poitevin, S.; Sallee, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef]
- Choi, E.Y.; Lee, H.; Dingle, R.W.; Kim, K.B.; Swanson, H.I. Development of novel CH223191-based antagonists of the aryl hydrocarbon receptor. Mol. Pharmacol. 2012, 81, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, C. Antioxidant Functions of the Aryl Hydrocarbon Receptor. Stem Cells Int. 2016, 2016, 7943495. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S.; Yamamoto, H.; Okamoto, A.; Imanishi, K.; Tokui, N.; Okamoto, T.; Suzuki, Y.; Sugiyama, N.; Imai, A.; Kudo, S.; et al. Effect of an Oral Adsorbent, AST-120, on Dialysis Initiation and Survival in Patients with Chronic Kidney Disease. Int. J. Nephrol. 2012, 2012, 376128. [Google Scholar] [CrossRef]
- Maeda, K.; Hamada, C.; Hayashi, T.; Shou, I.; Wakabayashi, M.; Fukui, M.; Horikoshi, S.; Tomino, Y. Long-term effects of the oral adsorbent, AST-120, in patients with chronic renal failure. J. Int. Med. Res. 2009, 37, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Shibahara, N.; Takagi, S.; Inoue, T.; Katsuoka, Y. AST-120, an oral adsorbent, delays the initiation of dialysis in patients with chronic kidney diseases. Ther. Apher. Dial. 2007, 11, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [Green Version]
- Kopf, P.G.; Huwe, J.K.; Walker, M.K. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc. Toxicol. 2008, 8, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, P.G.; Scott, J.A.; Agbor, L.N.; Boberg, J.R.; Elased, K.M.; Huwe, J.K.; Walker, M.K. Cytochrome P4501A1 is required for vascular dysfunction and hypertension induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2010, 117, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.H.; Yu, M.; Lee, S.; Ryu, D.R.; Kim, S.J.; Kang, D.H.; Choi, K.B. AST-120 Improves Microvascular Endothelial Dysfunction in End-Stage Renal Disease Patients Receiving Hemodialysis. Yonsei Med. J. 2016, 57, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, K.L.; Decker, E.; Perrenoud, L.; Kendrick, J.; Chonchol, M.; Seals, D.R.; Jalal, D. Assessment of vascular function in patients with chronic kidney disease. J. Vis. Exp. 2014, 88, e51478. [Google Scholar] [CrossRef]
- Lano, G.; Burtey, S.; Sallee, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Tumur, Z.; Niwa, T. Oral sorbent AST-120 increases renal NO synthesis in uremic rats. J. Ren. Nutr. 2008, 18, 60–64. [Google Scholar] [CrossRef]
- Eckers, A.; Jakob, S.; Heiss, C.; Haarmann-Stemmann, T.; Goy, C.; Brinkmann, V.; Cortese-Krott, M.M.; Sansone, R.; Esser, C.; Ale-Agha, N.; et al. The aryl hydrocarbon receptor promotes aging phenotypes across species. Sci. Rep. 2016, 6, 19618. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Liu, S.H.; Chiang, C.K.; Lin, S.Y.; Liang, K.W.; Chen, C.H.; Tien, H.R.; Chen, P.H.; Wu, J.P.; Tsai, Y.C.; et al. Aryl Hydrocarbon Receptor Deficiency Attenuates Oxidative Stress-Related Mesangial Cell Activation and Macrophage Infiltration and Extracellular Matrix Accumulation in Diabetic Nephropathy. Antioxid. Redox Signal. 2016, 24, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl sulfate, a uremic toxin, downregulates renal expression of Nrf2 through activation of NF-kappaB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Sallee, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Savira, F.; Kompa, A.R.; Magaye, R.; Xiong, X.; Huang, L.; Jucker, B.M.; Willette, R.N.; Kelly, D.J.; Wang, B.H. Apoptosis signal-regulating kinase 1 inhibition reverses deleterious indoxyl sulfate-mediated endothelial effects. Life Sci. 2021, 272, 119267. [Google Scholar] [CrossRef]
- Savira, F.; Magaye, R.; Scullino, C.V.; Flynn, B.L.; Pitson, S.M.; Anderson, D.; Creek, D.J.; Hua, Y.; Xiong, X.; Huang, L.; et al. Sphingolipid imbalance and inflammatory effects induced by uremic toxins in heart and kidney cells are reversed by dihydroceramide desaturase 1 inhibition. Toxicol. Lett. 2021, 350, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Yoo, T.H.; Cho, J.Y.; Kim, H.C.; Lee, W.W. Indoxyl sulfate-induced TNF-alpha is regulated by crosstalk between the aryl hydrocarbon receptor, NF-kappaB, and SOCS2 in human macrophages. FASEB J. 2019, 33, 10844–10858. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, J.; Ihara, K.; Nakayama, H.; Hikino, S.; Satoh, K.; Kubo, N.; Iida, T.; Fujii, Y.; Hara, T. Characteristic expression of aryl hydrocarbon receptor repressor gene in human tissues: Organ-specific distribution and variable induction patterns in mononuclear cells. Life Sci. 2004, 74, 1039–1049. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Kompa, A.R.; Manabe, M.; Wang, B.H.; Langham, R.G.; Nishijima, F.; Kelly, D.J.; Krum, H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS ONE 2012, 7, e41281. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S.; Kumfu, S.; Wang, B.H.; Manabe, M.; Nishijima, F.; Kelly, D.J.; Krum, H.; Kompa, A.R. The uremic toxin adsorbent AST-120 abrogates cardiorenal injury following myocardial infarction. PLoS ONE 2013, 8, e83687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namikoshi, T.; Tomita, N.; Satoh, M.; Sakuta, T.; Kuwabara, A.; Kobayashi, S.; Higuchi, Y.; Nishijima, F.; Kashihara, N. Oral adsorbent AST-120 ameliorates endothelial dysfunction independent of renal function in rats with subtotal nephrectomy. Hypertens. Res. 2009, 32, 194–200. [Google Scholar] [CrossRef]
- Ichii, O.; Otsuka-Kanazawa, S.; Nakamura, T.; Ueno, M.; Kon, Y.; Chen, W.; Rosenberg, A.Z.; Kopp, J.B. Podocyte injury caused by indoxyl sulfate, a uremic toxin and aryl-hydrocarbon receptor ligand. PLoS ONE 2014, 9, e108448. [Google Scholar] [CrossRef] [Green Version]
- Kompa, A.R.; Lu, J.; Weller, T.J.; Kelly, D.J.; Krum, H.; von Lueder, T.G.; Wang, B.H. Angiotensin receptor neprilysin inhibition provides superior cardioprotection compared to angiotensin converting enzyme inhibition after experimental myocardial infarction. Int. J. Cardiol. 2018, 258, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, J.; Tenkerian, C.; Gupta, J.; Ghaddar, N.; Wang, S.; Darini, C.; Staschke, K.A.; Ghosh, A.; Gandin, V.; Topisirovic, I.; et al. Downregulation of PERK activity and eIF2alpha serine 51 phosphorylation by mTOR complex 1 elicits pro-oxidant and pro-death effects in tuberous sclerosis-deficient cells. Cell Death Dis. 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aortic Ring Exposure Condition (N = 10) | -pEC50 (ACh) | Rmax (ACh) |
|---|---|---|
| Control | 7.34 ± 0.09 | 86.87 ± 3.52 |
| IS (300 μM) | 6.86 ± 0.12 ** | 50.31 ± 5.63 *** |
| IS + CH223191 (1 μM) | 7.16 ± 0.12 # | 77.49 ± 4.82 ### |
| IS + CH223191 (10 μM) | 7.26 ± 0.12 # | 81.48 ± 4.22 ### |
| Effect of CH223191 alone (N = 3) | ||
| Control | 7.31 ± 0.03 | 95.21 ± 1.16 |
| IS + CH223191 (1 μM) | 7.44 ± 0.23 | 87.34 ± 8.41 |
| IS + CH223191 (10 μM) | 7.05 ± 0.23 | 97.04 ± 1.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, C.; Edgley, A.J.; Kelly, D.J.; Kompa, A.R. Aryl Hydrocarbon Receptor Inhibition Restores Indoxyl Sulfate-Mediated Endothelial Dysfunction in Rat Aortic Rings. Toxins 2022, 14, 100. https://doi.org/10.3390/toxins14020100
Nguyen C, Edgley AJ, Kelly DJ, Kompa AR. Aryl Hydrocarbon Receptor Inhibition Restores Indoxyl Sulfate-Mediated Endothelial Dysfunction in Rat Aortic Rings. Toxins. 2022; 14(2):100. https://doi.org/10.3390/toxins14020100
Chicago/Turabian StyleNguyen, Cindy, Amanda J. Edgley, Darren J. Kelly, and Andrew R. Kompa. 2022. "Aryl Hydrocarbon Receptor Inhibition Restores Indoxyl Sulfate-Mediated Endothelial Dysfunction in Rat Aortic Rings" Toxins 14, no. 2: 100. https://doi.org/10.3390/toxins14020100
APA StyleNguyen, C., Edgley, A. J., Kelly, D. J., & Kompa, A. R. (2022). Aryl Hydrocarbon Receptor Inhibition Restores Indoxyl Sulfate-Mediated Endothelial Dysfunction in Rat Aortic Rings. Toxins, 14(2), 100. https://doi.org/10.3390/toxins14020100