Programmed Aptamer Screening, Characterization, and Rapid Detection for α-Conotoxin MI


 ,
,
Abstract
1. Introduction
2. Results and Discussion
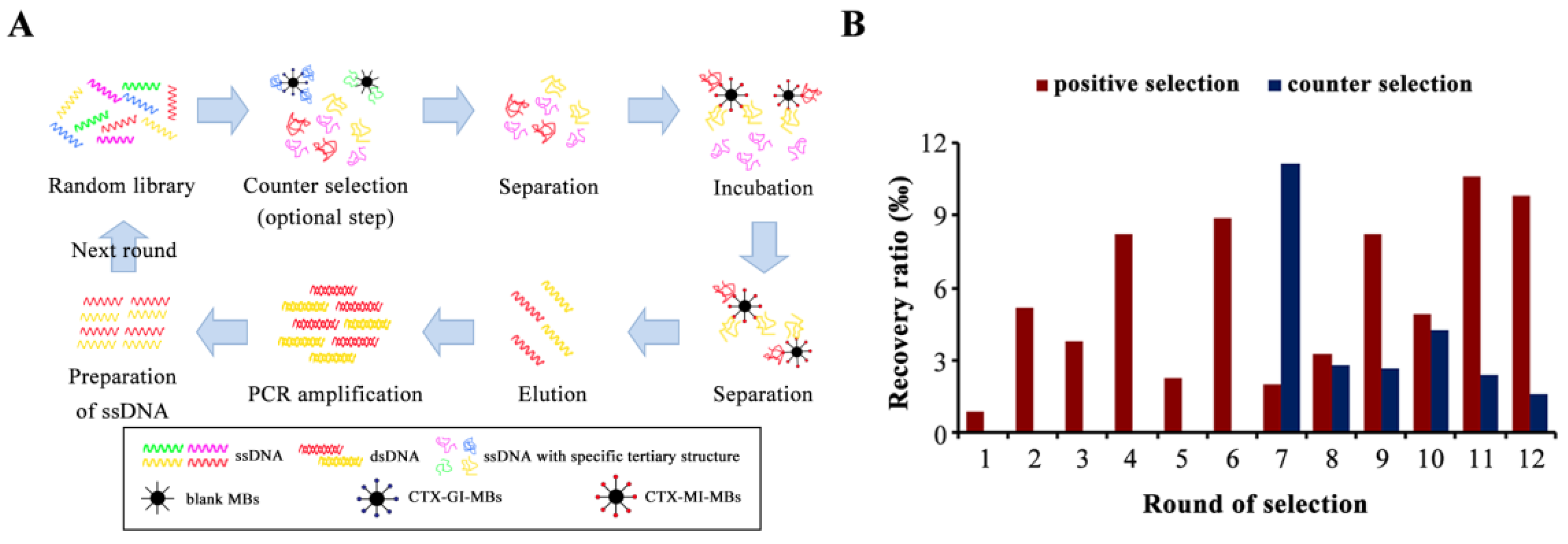
2.1. The Isolation of Aptamers for CTX-MI
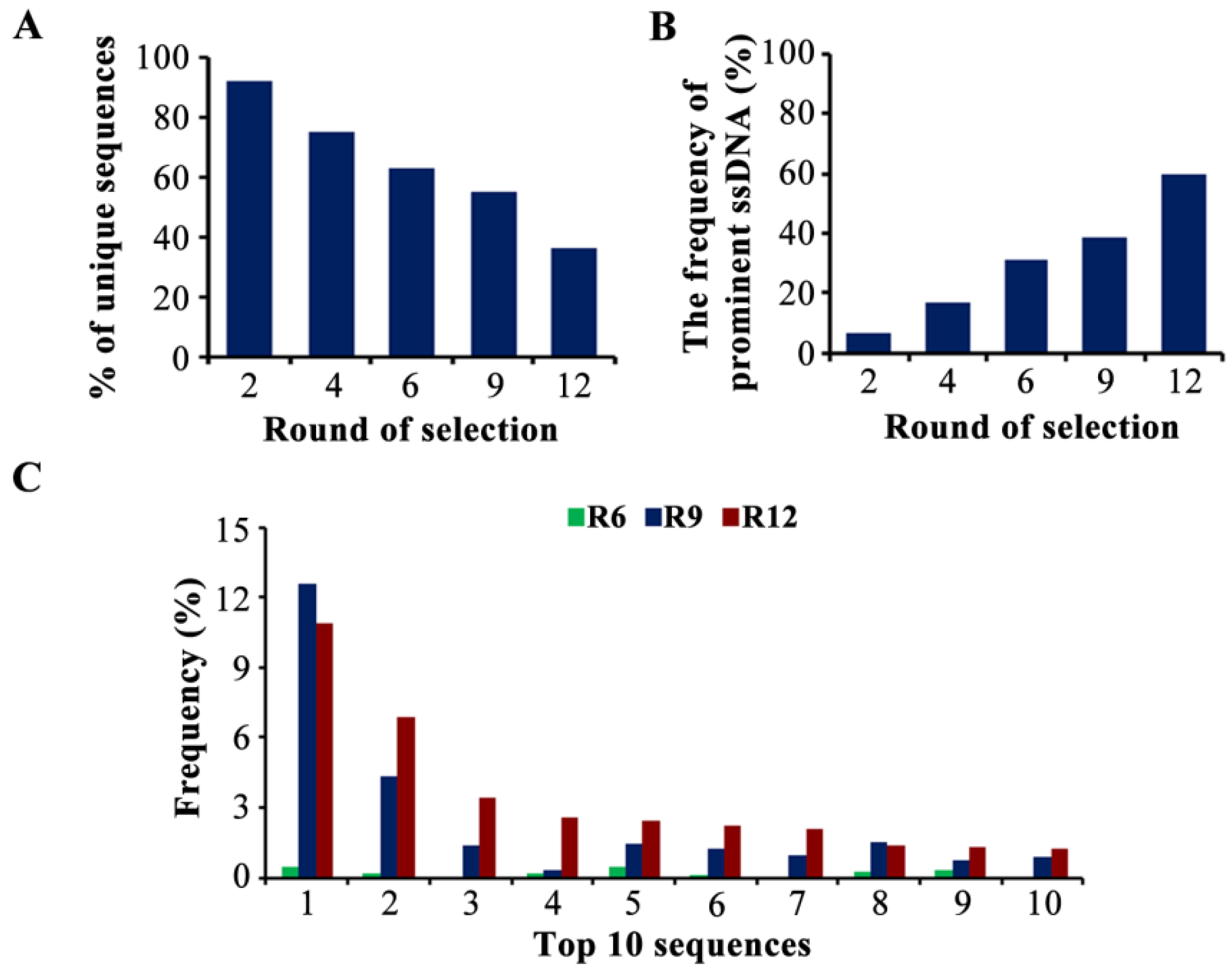
2.2. The Selection of Candidate Aptamers
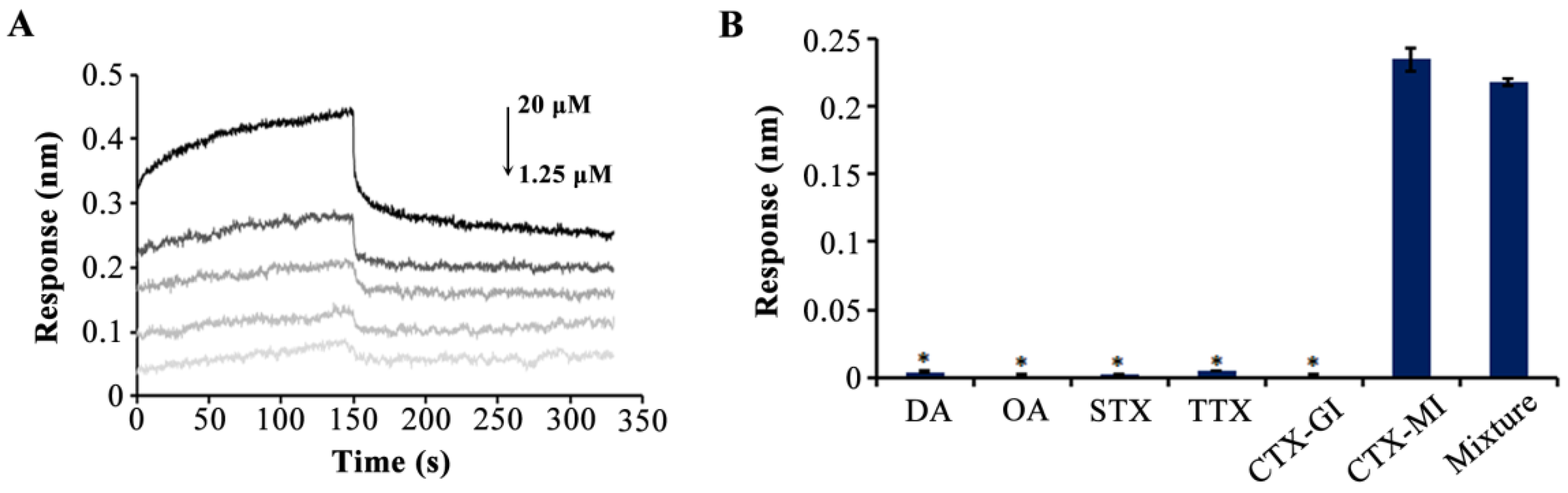
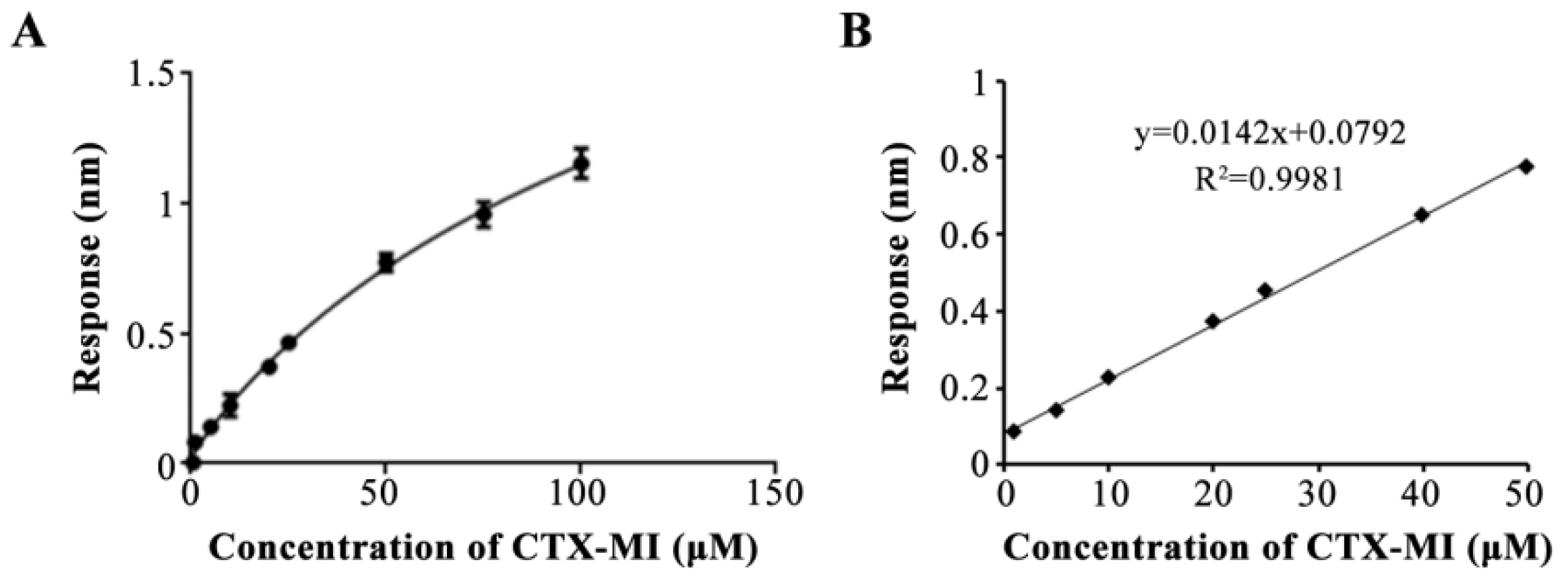
2.3. The Determination of Affinity and Specificity
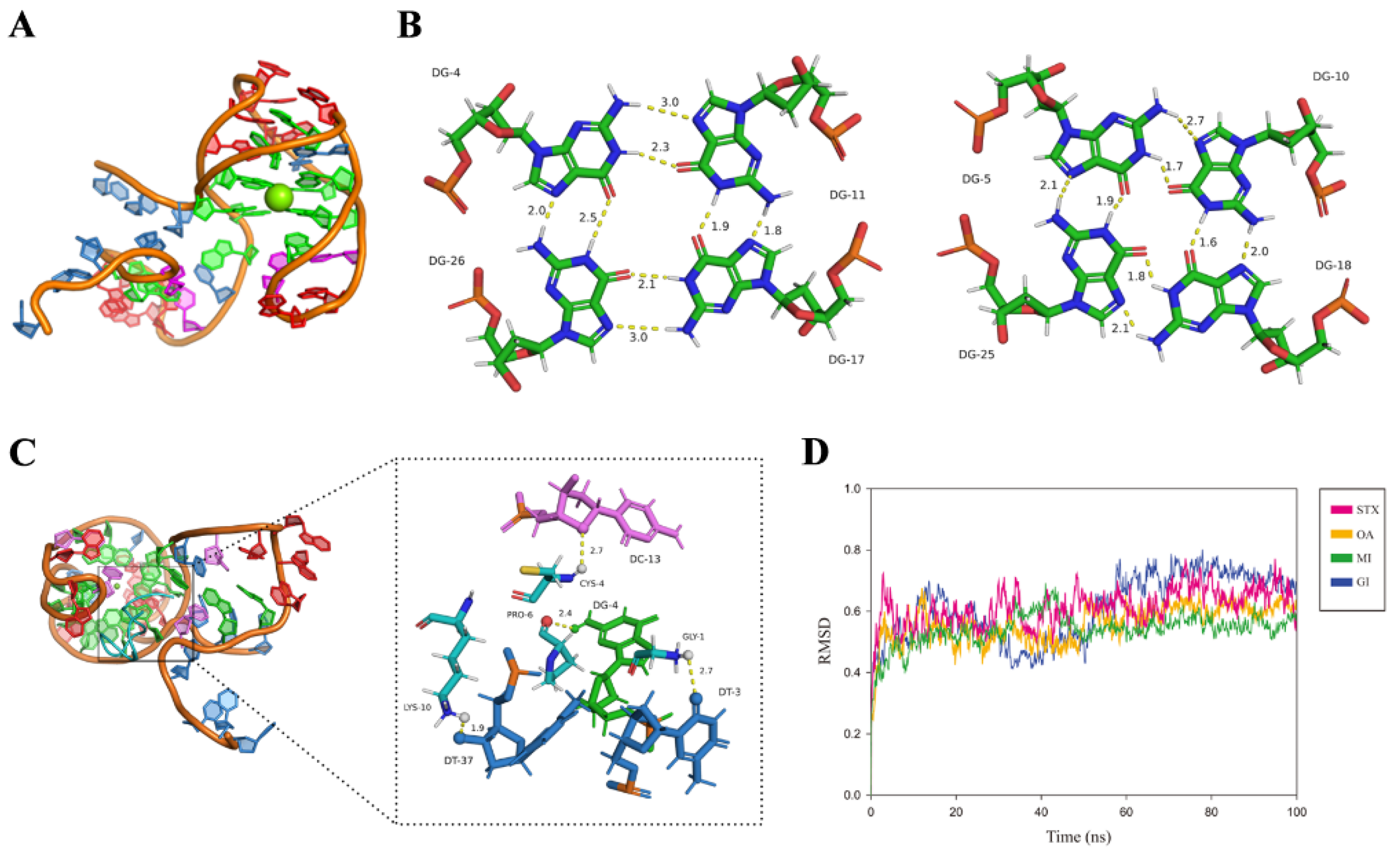
2.4. The Interaction Mechanism between MBMI-01c and CTX-MI
2.5. The Development of a BLI-Based Aptasensor for CTX-MI Detection
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Aptamers Selection In Vitro
4.2.1. Preparation of MBs
4.2.2. Random ssDNA Library and Primers
4.2.3. Aptamer Selection by MB-SELEX
4.3. The Preparation of HTS Samples
4.4. The Determination of Affinity by BLI Assay
4.5. Computer Simulation
4.5.1. Three-Dimensional Structure Prediction of MBMI-01c
4.5.2. Molecular Docking
4.5.3. MD Simulation
4.6. Performance Evaluation of the BLI Aptasensor
4.7. Treatment of Real Samples
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| BLI | biolayer interferometry |
| CTX-GI | α-conotoxin GI |
| CTX-GI-MBs | CTX-GI coated MBs |
| CTX-MI | α-conotoxin MI |
| CTX-MI-MBs | CTX-MI coated MBs |
| CTXs | conotoxins |
| CV | coefficient of variation |
| DA | domoic acid |
| GQ | G-quadruplet |
| HPLC | high-performance liquid chromatography |
| HTS | high-throughput sequencing |
| LOD | limit of detection |
| LOQ | limit of quantification |
| MALDI | matrix-assisted laser desorption/ionization |
| MB | magnetic beads |
| MD | molecular dynamics |
| MS | mass spectrometry |
| N-J tree | neighbor-joining tree |
| OA | okadaic acid |
| PME | Particle-Mesh-Ewald |
| QGRS prediction | G-rich sequences prediction |
| RMSD | root mean square deviation |
| SB | selection buffer |
| SELEX | systematic evolution of ligands by exponential enrichment |
| SSA | super streptavidin |
| STX | saxitoxin |
| TdMD | temperature-dependent molecular dynamics simulation |
| TTX | tetrodotoxin |
| 3D | three-dimensional |
References
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef]
- Becker, S.; Terlau, H. Toxins from cone snails: Properties, applications and biotechnological production. Appl. Microbiol. Biotechnol. 2008, 79, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Yao, G.; Gao, B.M.; Fan, C.X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. Gigascience 2016, 5, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.; Jin, A.H.; Hamilton, B.; Rai, S.K.; Alewood, P.; Lewis, R.J. Venom duct origins of prey capture and defensive conotoxins in piscivorous Conus striatus. Sci. Rep. 2021, 11, 13282–13295. [Google Scholar] [CrossRef]
- Robinson, S.D.; Norton, R.S. Conotoxin gene superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Gowd, K.H.; Sabareesh, V.; Sudarslal, S.; Iengar, P.; Franklin, B.; Fernando, A.; Dewan, K.; Ramaswami, M.; Sarma, S.P.; Sikdar, S.; et al. Novel peptides of therapeutic promise from Indian Conidae. Ann. N. Y. Acad. Sci. 2005, 1056, 462–473. [Google Scholar] [CrossRef]
- Kohn, A.J.; Saunders, P.R.; Wiener, S. Preliminary studies on the venom of the marine snail Conus. Ann. N. Y. Acad. Sci. 1960, 90, 706–725. [Google Scholar] [CrossRef]
- Rice, R.D.; Halstead, B.W. Report of fatal cone shell sting by Conus geographus Linnaeus. Toxicon 1968, 5, 223–224. [Google Scholar] [CrossRef]
- Endean, R.; Rudkin, C. Further studies of the venoms of conidae. Toxicon 1965, 2, 225–249. [Google Scholar] [CrossRef]
- Gray, W.R.; Rivier, J.E.; Galyean, R.; Cruz, L.J.; Olivera, B.M. Conotoxin MI. Disulfide bonding and conformational states. J. Biol. Chem. 1983, 258, 12247–12251. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, S.; Zhang, X.; Huang, Q.; Huang, Y.; Luo, M.; Wei, Y.; Chen, W.; Chen, Z.; Zhou, X.; et al. A new protein-coupled antigen of alpha-conotoxin MI displays high immunogenicity and can produce antiserum with high detoxification activity. Toxicon 2022, 208, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Shigeri, Y.; Inazumi, S.; Hagihara, Y.; Yasuda, A.; Kawasaki, H.; Arakawa, R.; Nakata, M. Desorption/ionization efficiency of peptides containing disulfide bonds in matrix-assisted laser desorption/ionization mass spectrometry. Anal. Sci. 2012, 28, 295–299. [Google Scholar] [CrossRef]
- Dionysiou, D. Overview: Harmful algal blooms and natural toxins in fresh and marine waters—Exposure, occurrence, detection, toxicity, control, management and policy. Toxicon 2010, 55, 907–908. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent advances in aptamer discovery and applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef]
- Ye, W.; Liu, T.; Zhang, W.; Zhu, M.; Liu, Z.; Kong, Y.; Liu, S. Marine toxins detection by biosensors based on aptamers. Toxins 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Jayasena, S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar] [CrossRef]
- Gao, S.; Hu, B.; Zheng, X.; Cao, Y.; Liu, D.; Sun, M.; Jiao, B.; Wang, L. Gonyautoxin 1/4 aptamers with high-affinity and high-specificity: From efficient selection to aptasensor application. Biosens. Bioelectron. 2016, 79, 938–944. [Google Scholar] [CrossRef]
- Kaur, H.; Shorie, M.; Sabherwal, P. Biolayer interferometry-SELEX for Shiga toxin antigenic-peptide aptamers & detection via chitosan-WSe2 aptasensor. Biosens. Bioelectron. 2020, 167, 112498–112522. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Anand, A.; Jain, N.; Goswami, S.; Anantharaj, A.; Patil, S.; Singh, R.; Kumar, A.; Shrivastava, T.; Bhatnagar, S.; et al. A novel G-quadruplex aptamer-based spike trimeric antigen test for the detection of SARS-CoV-2. Mol. Ther.-Nucleic Acids 2021, 26, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Shkembi, X.; Skouridou, V.; Svobodova, M.; Leonardo, S.; Bashammakh, A.S.; Alyoubi, A.O.; Campàs, M.; O’ Sullivan, C.K. Hybrid antibody—Aptamer assay for detection of tetrodotoxin in pufferfish. Anal. Chem. 2021, 93, 14810–14819. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M. Purification and properties of a myotoxin from Conus geographus venom. Arch. Biochem. Biophys. 1978, 190, 539–548. [Google Scholar] [CrossRef]
- Mergny, J.L.; Phan, A.T.; Lacroix, L. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998, 435, 74–78. [Google Scholar] [CrossRef]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5’-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Huppert, J.L. Four-stranded DNA: Cancer, gene regulation and drug development. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2007, 365, 2969–2984. [Google Scholar] [CrossRef]
- Oganesian, L.; Bryan, T.M. Physiological relevance of telomeric G-quadruplex formation: A potential drug target. Bioessays 2007, 29, 155–165. [Google Scholar] [CrossRef]
- Wang, K.Y.; McCurdy, S.; Shea, R.G.; Swaminathan, S.; Bolton, P.H. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry 1993, 32, 1899–1904. [Google Scholar] [CrossRef]
- Marsh, T.C.; Vesenka, J.; Henderson, E. A new DNA nanostructure, the G-wire, imaged by scanning probe microscopy. Nucleic Acids Res. 1995, 23, 696–700. [Google Scholar] [CrossRef]
- Song, M.; Li, G.; Zhang, Q.; Liu, J.; Huang, Q. De novo post-SELEX optimization of a G-quadruplex DNA aptamer binding to marine toxin gonyautoxin 1/4. Comput. Struct. Biotechnol. J. 2020, 18, 3425–3433. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.; Witte, K.; Wartchow, C.; Choo, S.; Yao, D.; Persson, H.; Wei, J.; Li, P.; Heidecker, B.; Ma, W.; et al. Label-free detection of biomolecular interactions using BioLayer interferometry for kinetic characterization. Comb. Chem. High Throughput Screen. 2009, 12, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Lee, J.E. Measuring protein-protein and protein-nucleic acid interactions by biolayer interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Hu, B.; Zhou, R.; Liu, D.; Peng, D.; Li, Z.; Li, Z.; Jiao, B.; Wang, L. Rapid and sensitive detection of nodularin-R in water by a label-free BLI aptasensor. Analyst 2018, 143, 4316–4322. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, Y.; Deng, B.; Hu, B.; Zhao, L.; Guo, H.; Yang, C.; Ma, Z.; Sun, M.; Jiao, B.; et al. Selection, characterization, and optimization of DNA aptamers against challenging marine biotoxin gymnodimine-a for biosensing application. Toxins 2022, 14, 195. [Google Scholar] [CrossRef]
- Bren, N.; Sine, S.M. Hydrophobic pairwise interactions stabilize alpha-conotoxin MI in the muscle acetylcholine receptor binding site. J. Biol. Chem. 2000, 275, 12692–12700. [Google Scholar] [CrossRef]
- Wong, H.M.; Stegle, O.; Rodgers, S.; Huppert, J.L. A toolbox for predicting g-quadruplex formation and stability. J. Nucleic Acids 2010, 2010, 564946. [Google Scholar] [CrossRef]
- Hu, B.; Zhou, R.; Li, Z.; Ouyang, S.; Li, Z.; Hu, W.; Wang, L.; Jiao, B. Study of the binding mechanism of aptamer to palytoxin by docking and molecular simulation. Sci. Rep. 2019, 9, 15494–15504. [Google Scholar] [CrossRef]
- Mufhandu, H.T.; Gray, E.S.; Madiga, M.C.; Tumba, N.; Alexandre, K.B.; Khoza, T.; Wibmer, C.K.; Moore, P.L.; Morris, L.; Khati, M. UCLA1, a synthetic derivative of a gp120 RNA aptamer, inhibits entry of human immunodeficiency virus type 1 subtype C. J. Virol. 2012, 86, 4989–4999. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure–activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L.; et al. Novel omega-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar] [CrossRef] [PubMed]
- Livett, B.G.; Gayler, K.R.; Khalil, Z. Drugs from the sea: Conopeptides as potential therapeutics. Curr. Med. Chem. 2004, 11, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Livett, B.G.; Sandall, D.W.; Keays, D.; Down, J.; Gayler, K.R.; Satkunanathan, N.; Khalil, Z. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 2006, 48, 810–829. [Google Scholar] [CrossRef] [PubMed]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Patro, L.; Kumar, A.; Kolimi, N.; Rathinavelan, T. 3D-NuS: A web server for automated modeling and visualization of non-canonical 3-dimensional nucleic acid structures. J. Mol. Biol. 2017, 429, 2438–2448. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 2010, 4, 187–217. [Google Scholar] [CrossRef]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.R.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.J.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Henrik, G.P. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Shrivastava, A. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Drug Discov. Ther. 2011, 2, 21–25. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Etotal (kcal/mol) | Eshape (kcal/mol) | Eforce (kcal/mol) | RMS |
|---|---|---|---|---|
| CTX-MI | −531.44 | −531.44 | 0 | −1.00 |
| CTX-GI | −439.61 | −439.61 | 0 | −1.00 |
| OA | −264.49 | −264.49 | 0 | −1.00 |
| STX | −388.01 | −388.01 | 0 | −1.00 |
| CTX-MI (μM) | Detection Value (μM) | CV (%) |
|---|---|---|
| 5 | 5.48 | 3.25 |
| 10 | 10.21 | 2.45 |
| 20 | 20.14 | 2.00 |
| CTX-MI (μM) | Detection Value (μM) | CV (%) | Recovery (%) |
|---|---|---|---|
| 10 | 10.47 | 4.31 | 102.11 |
| 25 | 24.71 | 4.90 | 98.83 |
| 50 | 50.53 | 2.21 | 101.06 |
| ID | Sequence (5′ to 3′) |
|---|---|
| Lib V1 | ATTGGCACTCCACGCATAGG-N40-CCTATGCGTGCTACCGTGAA |
| F1 | ATTGGCACTCCACGCATAGG |
| F2 | AAAGCAATTGGCACTCCACGCATAGG |
| F3 | AACGCCATTGGCACTCCACGCATAGG |
| F4 | AAGGCGATTGGCACTCCACGCATAGG |
| F5 | ACAGGAATTGGCACTCCACGCATAGG |
| F6 | ACCGGCATTGGCACTCCACGCATAGG |
| R1 | A20-Spacer18-TTCACGGTAGCACGCATAGG |
| R2 | TTCACGGTAGCACGCATAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Deng, B.; Zhao, L.; Gao, Y.; Zhang, X.; Yang, C.; Zou, B.; Chen, H.; Sun, M.; Wang, L.; et al. Programmed Aptamer Screening, Characterization, and Rapid Detection for α-Conotoxin MI. Toxins 2022, 14, 706. https://doi.org/10.3390/toxins14100706
Guo H, Deng B, Zhao L, Gao Y, Zhang X, Yang C, Zou B, Chen H, Sun M, Wang L, et al. Programmed Aptamer Screening, Characterization, and Rapid Detection for α-Conotoxin MI. Toxins. 2022; 14(10):706. https://doi.org/10.3390/toxins14100706
Chicago/Turabian StyleGuo, Han, Bowen Deng, Luming Zhao, Yun Gao, Xiaojuan Zhang, Chengfang Yang, Bin Zou, Han Chen, Mingjuan Sun, Lianghua Wang, and et al. 2022. "Programmed Aptamer Screening, Characterization, and Rapid Detection for α-Conotoxin MI" Toxins 14, no. 10: 706. https://doi.org/10.3390/toxins14100706
APA StyleGuo, H., Deng, B., Zhao, L., Gao, Y., Zhang, X., Yang, C., Zou, B., Chen, H., Sun, M., Wang, L., & Jiao, B. (2022). Programmed Aptamer Screening, Characterization, and Rapid Detection for α-Conotoxin MI. Toxins, 14(10), 706. https://doi.org/10.3390/toxins14100706