Human Peptides α-Defensin-1 and -5 Inhibit Pertussis Toxin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
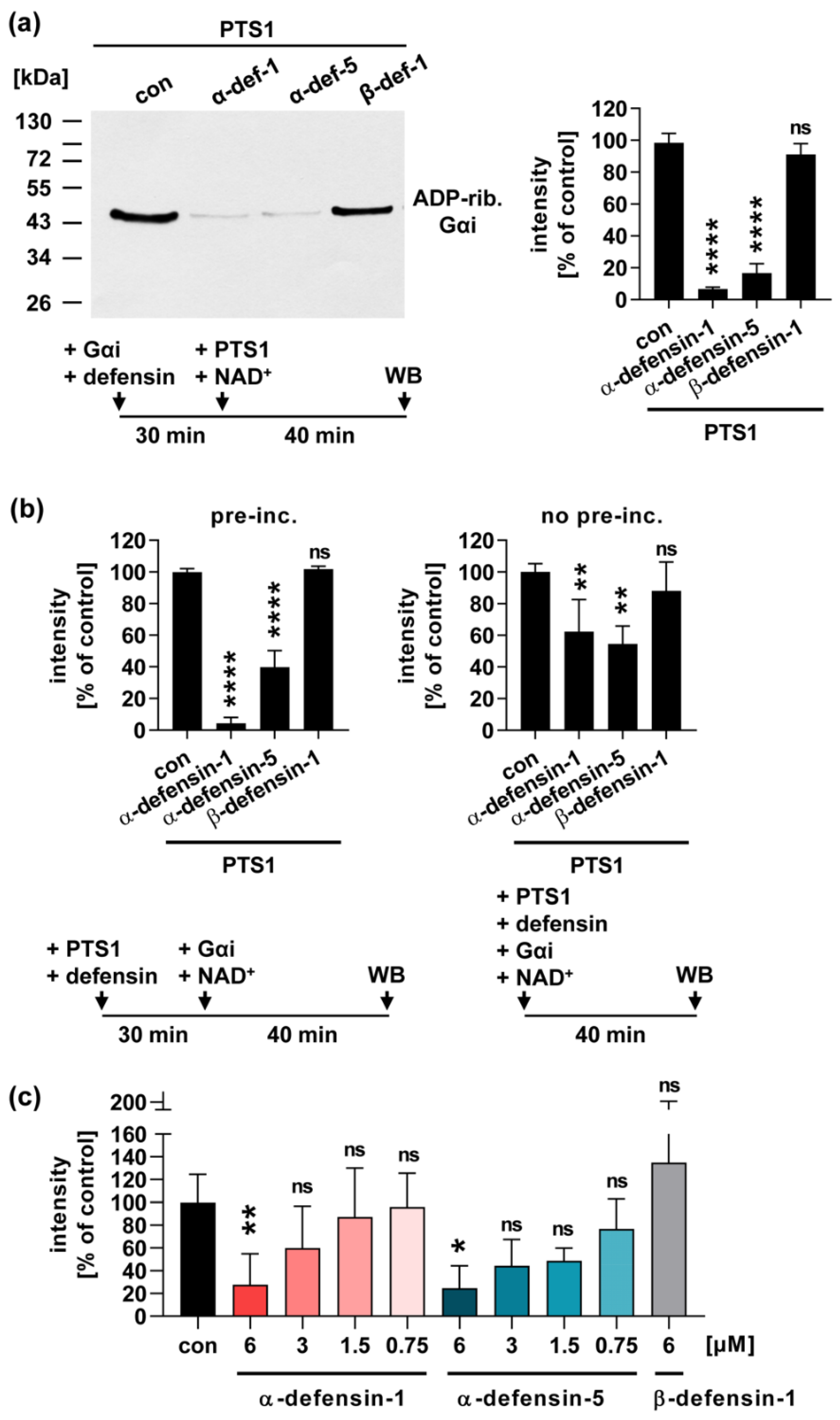
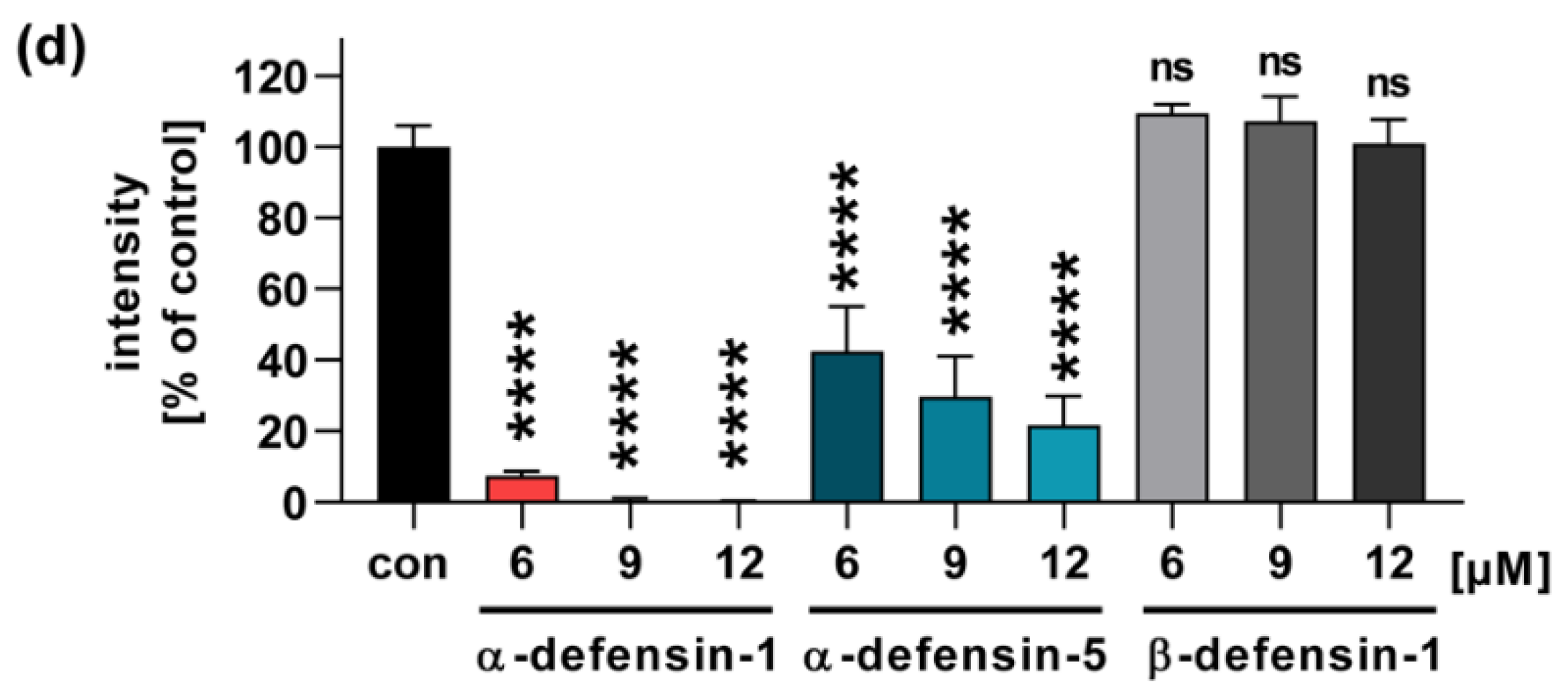
2.1. Human α-Defensin-1 and -5 Inhibit the Enzyme Activity of PTS1 In Vitro
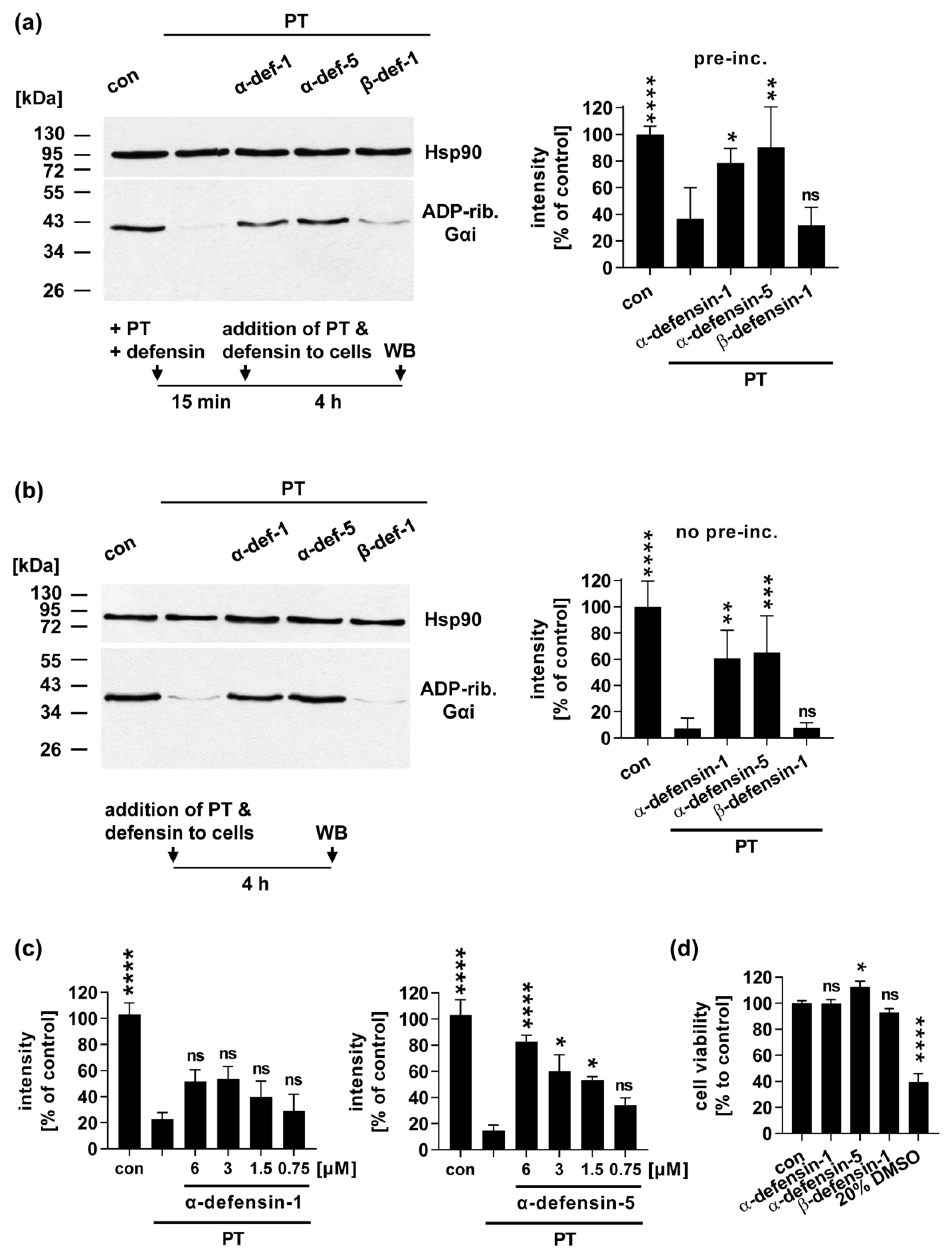
2.2. In the Presence of α-Defensin-1 and -5 Less Gαi Is ADP-Ribosylated in PT-Treated Cells
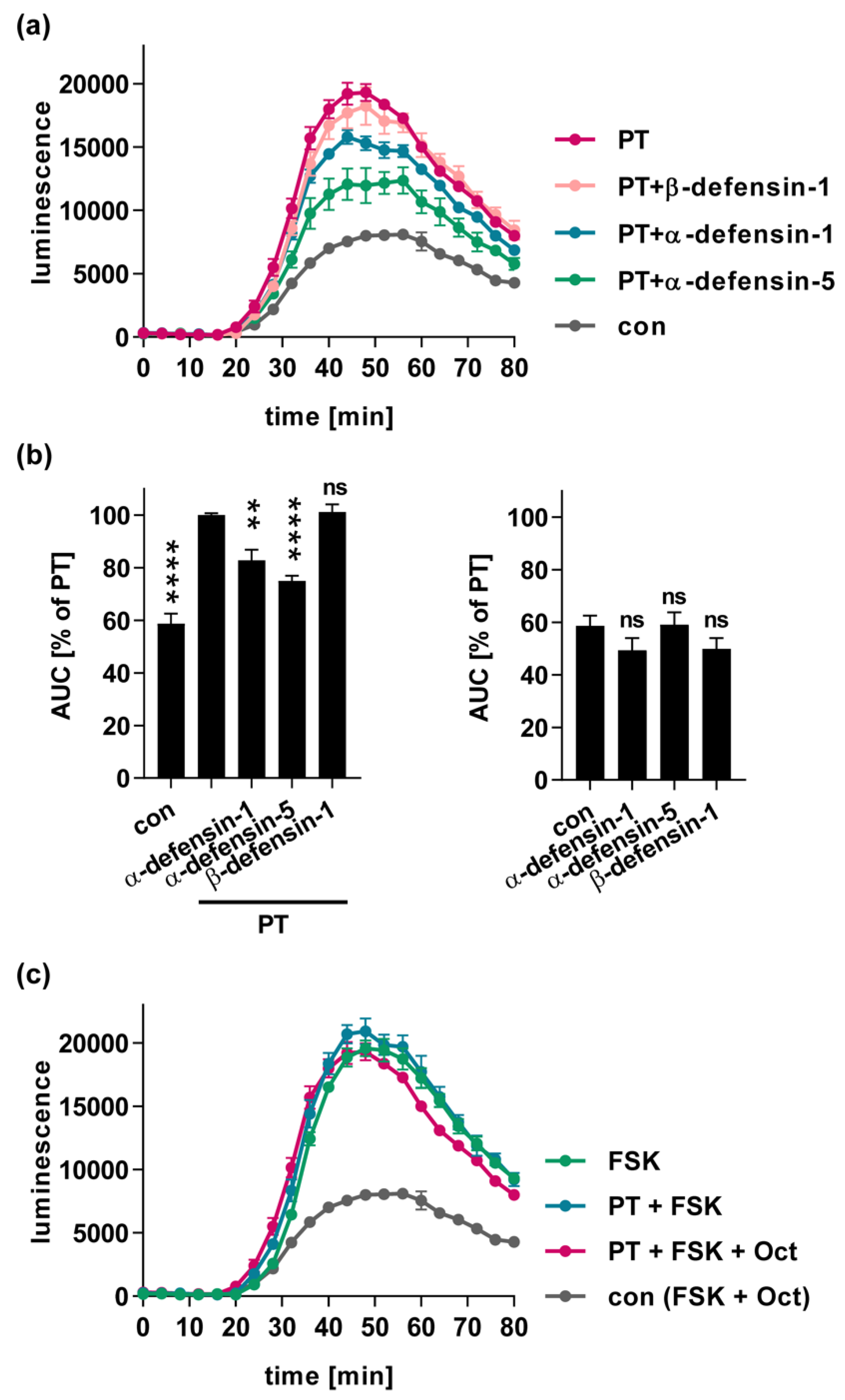
2.3. α-Defensin-1 and -5 Inhibit the PT-Mediated Effects on cAMP Signaling in Cells
3. Discussion
4. Materials and Methods
4.1. Compounds and Reagents
4.2. In vitro Enzyme Activity of PTS1
4.3. Cell Lines
4.4. Sequential ADP-Ribosylation of Gαi in Lysates from Toxin-Treated Cells
4.5. Cell Viability Assay
4.6. iGIST Bioassay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Tamura, M.; Nogimori, K.; Murai, S.; Yajima, M.; Ito, K.; Katada, T.; Ui, M.; Ishii, S. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry 1982, 21, 5516–5522. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.A.; Johnson, F.D.; Burns, D.L. Molecular characterization of an operon required for pertussis toxin secretion. Proc. Natl. Acad. Sci. USA 1993, 90, 2970–2974. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, G.D.; Howard, L.A.; Peppler, M.S. Use of glycosyltransferases to restore pertussis toxin receptor activity to asialoagalactofetuin. J. Biol. Chem. 1988, 263, 8677–8684. [Google Scholar] [CrossRef]
- Hausman, S.Z.; Burns, D.L. Binding of pertussis toxin to lipid vesicles containing glycolipids. Infect. Immun. 1993, 61, 335–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witvliet, M.H.; Burns, D.L.; Brennan, M.J.; Poolman, J.T.; Manclark, C.R. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect. Immun. 1989, 57, 3324–3330. [Google Scholar] [CrossRef] [Green Version]
- El Bayâ, A.; Linnemann, R.; Von Olleschik-Elbheim, L.; Robenek, H.; Schmidt, M.A. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur. J. Cell Biol. 1997, 73, 40–48. [Google Scholar]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid Redistribution of Golgi Proteins into the ER in Cells Treated with Brefeldin A: Evidence for Membrane Cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Burns, D.L.; Manclark, C.R. Adenine nucleotides promote dissociation of pertussis toxin subunits. J. Biol. Chem. 1986, 261, 4324–4327. [Google Scholar] [CrossRef]
- Hazes, B.; Boodhoo, A.; Cockle, S.A.; Read, R. Crystal Structure of the Pertussis Toxin–ATP Complex: A Molecular Sensor. J. Mol. Biol. 1996, 258, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaut, R.; Scanlon, K.M.; Taylor, M.; Teter, K.; Carbonetti, N.H. Intracellular disassembly and activity of pertussis toxin require interaction with ATP. Pathog. Dis. 2016, 74, 74. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Cilenti, L.; Taylor, M.; Showman, A.; Tatulian, S.A.; Teter, K. Thermal Unfolding of the Pertussis Toxin S1 Subunit Facilitates Toxin Translocation to the Cytosol by the Mechanism of Endoplasmic Reticulum-Associated Degradation. Infect. Immun. 2016, 84, 3388–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazes, B.; Read, R. Accumulating Evidence Suggests That Several AB-Toxins Subvert the Endoplasmic Reticulum-Associated Protein Degradation Pathway To Enter Target Cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.H.; Moe, D.; Jamnadas, M.; Tatulian, S.A.; Teter, K. The Pertussis Toxin S1 Subunit Is a Thermally Unstable Protein Susceptible to Degradation by the 20S Proteasome†. Biochemistry 2006, 45, 13734–13740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthington, Z.E.V.; Carbonetti, N.H. Evading the Proteasome: Absence of Lysine Residues Contributes to Pertussis Toxin Activity by Evasion of Proteasome Degradation. Infect. Immun. 2007, 75, 2946–2953. [Google Scholar] [CrossRef] [Green Version]
- Ernst, K.; Eberhardt, N.; Mittler, A.-K.; Sonnabend, M.; Anastasia, A.; Freisinger, S.; Schiene-Fischer, C.; Malešević, M.; Barth, H. Pharmacological Cyclophilin Inhibitors Prevent Intoxication of Mammalian Cells with Bordetella pertussis Toxin. Toxins 2018, 10, 181. [Google Scholar] [CrossRef] [Green Version]
- Ernst, K.; Mittler, A.-K.; Winkelmann, V.; Kling, C.; Eberhardt, N.; Anastasia, A.; Sonnabend, M.; Lochbaum, R.; Wirsching, J.; Sakari, M.; et al. Pharmacological targeting of host chaperones protects from pertussis toxin in vitro and in vivo. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Bokoch, G.M.; Katada, T.; Northup, J.K.; Hewlett, E.L.; Gilman, A.G. Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J. Biol. Chem. 1983, 258, 2072–2075. [Google Scholar] [CrossRef]
- Katada, T.; Ui, M. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc. Natl. Acad. Sci. USA 1982, 79, 3129–3133. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, C.; Carbonetti, N.H. Pertussis Toxin Inhibits Early Chemokine Production to Delay Neutrophil Recruitment in Response to Bordetella pertussis Respiratory Tract Infection in Mice. Infect. Immun. 2008, 76, 5139–5148. [Google Scholar] [CrossRef] [Green Version]
- Kirimanjeswara, G.S.; Agosto, L.M.; Kennett, M.J.; Bjornstad, O.N.; Harvill, E.T. Pertussis toxin inhibits neutrophil recruitment to delay antibody-mediated clearance of Bordetella pertussis. J. Clin. Investig. 2005, 115, 3594–3601. [Google Scholar] [CrossRef] [Green Version]
- Spangrude, G.J.; Sacchi, F.; Hill, H.R.; Van Epps, D.E.; Daynes, R.A. Inhibition of lymphocyte and neutrophil chemotaxis by pertussis toxin. J. Immunol. 1985, 135, 4135–4143. [Google Scholar]
- Mattoo, S.; Cherry, J.D. Molecular Pathogenesis, Epidemiology, and Clinical Manifestations of Respiratory Infections Due to Bordetella pertussis and Other Bordetella Subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An update of the global burden of pertussis in children younger than 5 years: A modelling study. Lancet Infect. Dis. 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Robert-Koch-Institut. Impfquoten bei der Schuleingangsuntersuchung in Deutschland 2017. Epid Bull 2019, 18, 147–153. [Google Scholar] [CrossRef]
- WHO. Immunization Coverage. Available online: https://www.who.int/news-room/fact-sheets/detail/immunization-coverage (accessed on 7 June 2021).
- Connelly, C.E.; Sun, Y.; Carbonetti, N.H. Pertussis Toxin Exacerbates and Prolongs Airway Inflammatory Responses during Bordetella pertussis Infection. Infect. Immun. 2012, 80, 4317–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonetti, N.H. Contribution of pertussis toxin to the pathogenesis of pertussis disease: Graphical Abstract Figure. Pathog. Dis. 2015, 73, ftv073. [Google Scholar] [CrossRef]
- Pittman, M. The concept of pertussis as a toxin-mediated disease. Pediatr. Infect. Dis. J. 1984, 3, 467–486. [Google Scholar] [CrossRef]
- Scanlon, K.; Skerry, C.; Carbonetti, N. Association of Pertussis Toxin with Severe Pertussis Disease. Toxins 2019, 11, 373. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Slavinskaya, Z.; Merrill, A.R.; Kaufmann, S.H.E. Human α-defensins neutralize toxins of the mono-ADP-ribosyltransferase family. Biochem. J. 2006, 399, 225–229. [Google Scholar] [CrossRef]
- Kudryashova, E.; Seveau, S.M.; Kudryashov, D.S. Targeting and inactivation of bacterial toxins by human defensins. Biol. Chem. 2017, 398, 1069–1085. [Google Scholar] [CrossRef]
- Cederlund, A.; Gudmundsson, G.H.; Agerberth, B. Antimicrobial peptides important in innate immunity. FEBS J. 2011, 278, 3942–3951. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Ückert, A.; Landenberger, M.; Papatheodorou, P.; Hoffmann-Richter, C.; Mittler, A.; Ziener, U.; Hägele, M.; Schwan, C.; Müller, M.; et al. Human peptide α-defensin-1 interferes withClostridioides difficiletoxins TcdA, TcdB, and CDT. FASEB J. 2020, 34, 6244–6261. [Google Scholar] [CrossRef] [Green Version]
- Korbmacher, M.; Fischer, S.; Landenberger, M.; Papatheodorou, P.; Aktories, K.; Barth, H. Human α-Defensin-5 Efficiently Neutralizes Clostridioides difficile Toxins TcdA, TcdB, and CDT. Front. Pharmacol. 2020, 11, 1204. [Google Scholar] [CrossRef] [PubMed]
- Giesemann, T.; Guttenberg, G.; Aktories, K. Human α-Defensins Inhibit Clostridium difficile Toxin B. Gastroenterology 2008, 134, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Paramonov, V.M.; Sahlgren, C.; Rivero-Müller, A.; Pulliainen, A.T. iGIST—A Kinetic Bioassay for Pertussis Toxin Based on Its Effect on Inhibitory GPCR Signaling. ACS Sens. 2020, 5, 3438–3448. [Google Scholar] [CrossRef]
- Kilgore, P.E.; Salim, A.M.; Zervos, M.J.; Schmitt, H.-J. Pertussis: Microbiology, Disease, Treatment, and Prevention. Clin. Microbiol. Rev. 2016, 29, 449–486. [Google Scholar] [CrossRef] [Green Version]
- Kudryashova, E.; Quintyn, R.; Seveau, S.; Lu, W.; Wysocki, V.; Kudryashov, D.S. Human Defensins Facilitate Local Unfolding of Thermodynamically Unstable Regions of Bacterial Protein Toxins. Immunity 2014, 41, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kaufmann, S.H. Defensin: A multifunctional molecule lives up to its versatile name. Trends Microbiol. 2006, 14, 428–431. [Google Scholar] [CrossRef]
- Fischer, S.; Popoff, M.R.; Barth, H. Human alpha-defensin-1 protects cells from intoxication with Clostridium perfringens iota toxin. Pathog. Dis. 2018, 76, 76. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gajendran, N.; Mittrücker, H.-W.; Weiwad, M.; Song, Y.-H.; Hurwitz, R.; Wilmanns, M.; Fischer, G.; Kaufmann, S.H.E. Human -defensins neutralize anthrax lethal toxin and protect against its fatal consequences. Proc. Natl. Acad. Sci. USA 2005, 102, 4830–4835. [Google Scholar] [CrossRef] [Green Version]
- Sengeløv, H.; Follin, P.; Kjeldsen, L.; Lollike, K.; Dahlgren, C.; Borregaard, N.; Sengeløv, H.; Follin, P.; Kjeldsen, L.; Lollike, K.; et al. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J. Immunol. 1995, 154, 4157–4165. [Google Scholar] [PubMed]
- Eby, J.C.; Hoffman, C.; Gonyar, L.A.; Hewlett, E.L. Review of the neutrophil response toBordetella pertussisinfection. Pathog. Dis. 2015, 73, ftv081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.; Bevins, C. Paneth cells of the human small intestine express an antimicrobial peptide gene. J. Biol. Chem. 1992, 267, 23216–23225. [Google Scholar] [CrossRef]
- Ashok, Y.; Miettinen, M.; de Oliveira, D.K.H.; Tamirat, M.Z.; Näreoja, K.; Tiwari, A.; Hottiger, M.O.; Johnson, M.S.; Lehtiö, L.; Pulliainen, A.T. Discovery of Compounds Inhibiting the ADP-Ribosyltransferase Activity of Pertussis Toxin. ACS Infect. Dis. 2020, 6, 588–602. [Google Scholar] [CrossRef]
- Xu, Y.; Barbieri, J.T. Pertussis toxin-mediated ADP-ribosylation of target proteins in Chinese hamster ovary cells involves a vesicle trafficking mechanism. Infect. Immun. 1995, 63, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kling, C.; Pulliainen, A.T.; Barth, H.; Ernst, K. Human Peptides α-Defensin-1 and -5 Inhibit Pertussis Toxin. Toxins 2021, 13, 480. https://doi.org/10.3390/toxins13070480
Kling C, Pulliainen AT, Barth H, Ernst K. Human Peptides α-Defensin-1 and -5 Inhibit Pertussis Toxin. Toxins. 2021; 13(7):480. https://doi.org/10.3390/toxins13070480
Chicago/Turabian StyleKling, Carolin, Arto T. Pulliainen, Holger Barth, and Katharina Ernst. 2021. "Human Peptides α-Defensin-1 and -5 Inhibit Pertussis Toxin" Toxins 13, no. 7: 480. https://doi.org/10.3390/toxins13070480
APA StyleKling, C., Pulliainen, A. T., Barth, H., & Ernst, K. (2021). Human Peptides α-Defensin-1 and -5 Inhibit Pertussis Toxin. Toxins, 13(7), 480. https://doi.org/10.3390/toxins13070480