Comprehensive Metabolomic Analysis Reveals Dynamic Metabolic Reprogramming in Hep3B Cells with Aflatoxin B1 Exposure


Abstract
1. Introduction
2. Results
2.1. AFB1 Significantly Inhibits the Proliferation and Promotes the Apoptosis of Hep3B Cells
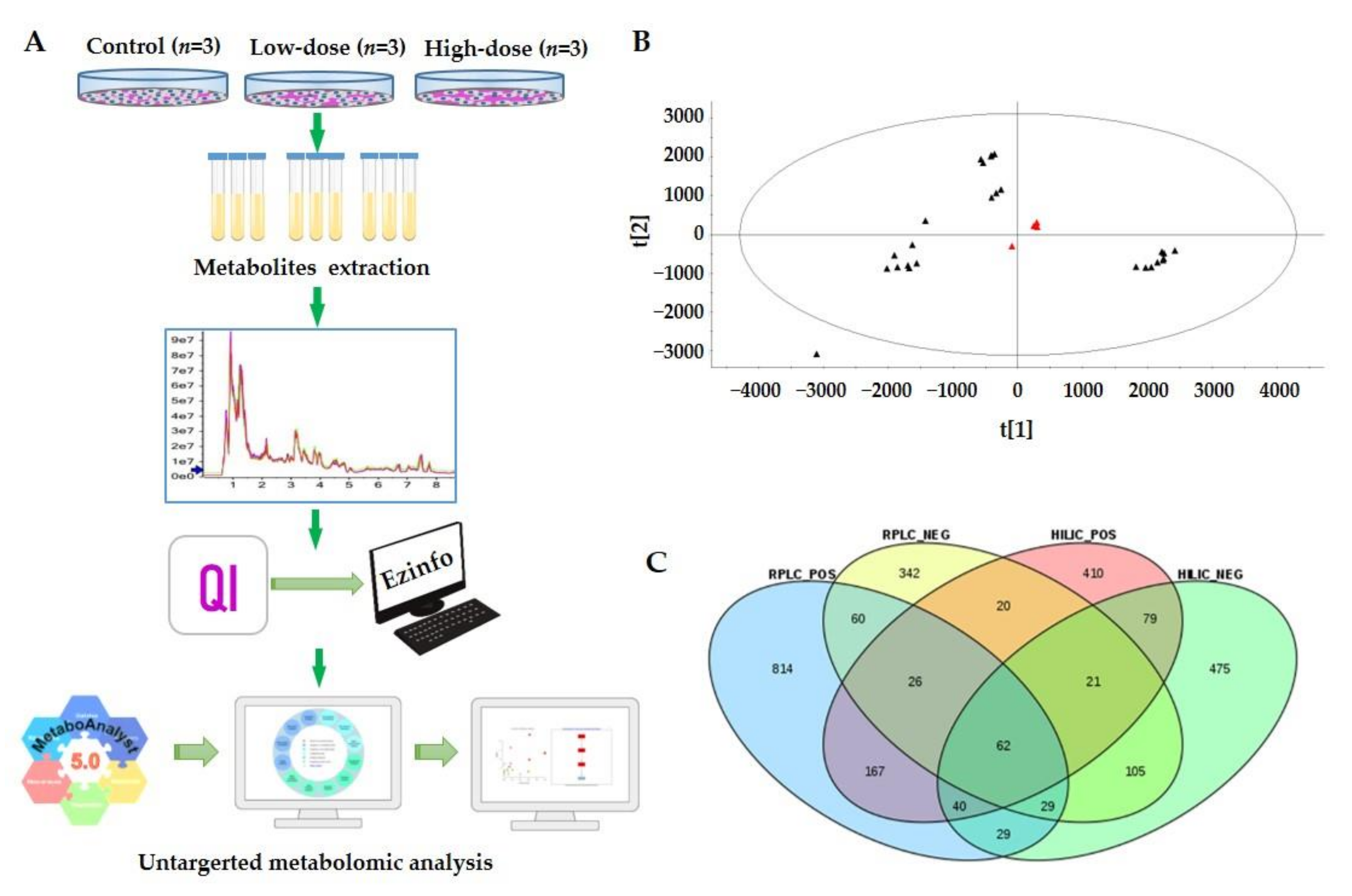
2.2. Metabolic Profiles Based on UPLC-ESI-MS/MS
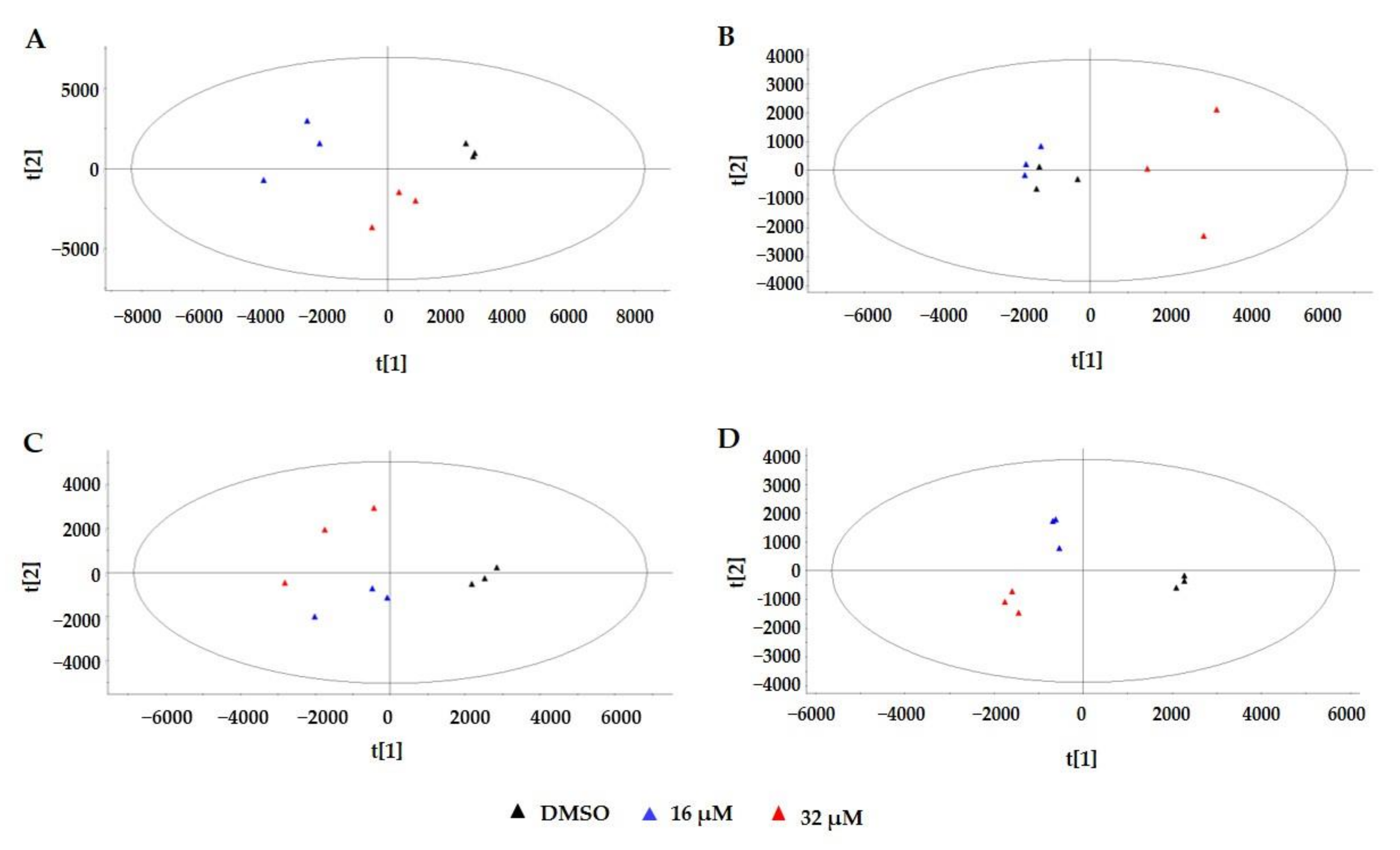
2.3. Multivariate Statistical Analysis
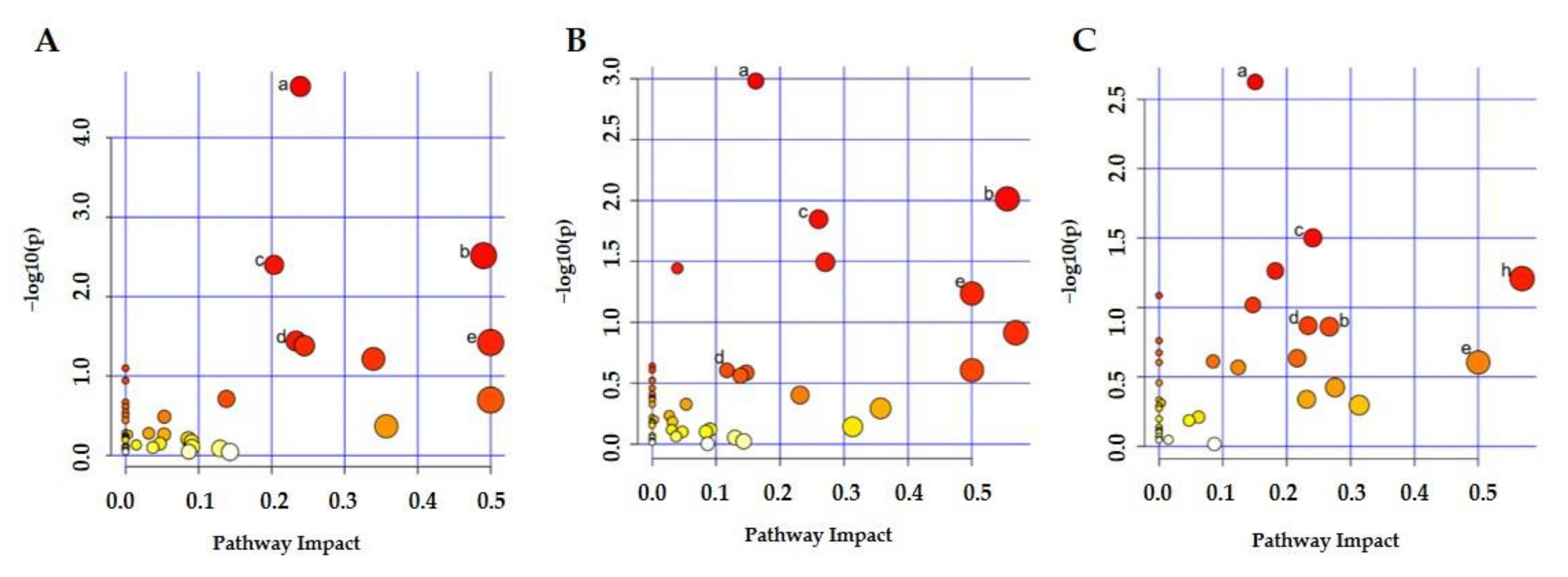
2.4. Metabolic Pathway Analysis
3. Discussion
3.1. Purine and Pyrimidine Metabolism
3.2. Lipid Metabolism and Oxidative Stress
3.3. Hexosamine Pathway and Sialylation
3.4. Amino Acid Metabolism
4. Conclusions
5. Materials and Methods
5.1. Instruments and Reagents
5.2. Experimental Method
5.2.1. Cell Treatment and Viability Assay
5.2.2. Flow Cytometry Analysis
5.2.3. Metabolite Extraction from Hep3B Cells
5.2.4. UPLC-ESI-MS/MS Analysis
5.2.5. Data Processing and Analysis
5.2.6. Metabolic Pathway Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Groopman, J.D.; Kensler, T.W.; Wild, C.P. Protective interventions to prevent aflatoxin-induced carcinogenesis in developing countries. Annu. Rev. Public Health 2008, 29, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Meissonnier, G.M.; Laffitte, J.; Loiseau, N.; Benoit, E.; Raymond, I.; Pinton, P.; Cossalter, A.-M.; Bertin, G.; Oswald, I.P.; Galtier, P. Selective impairment of drug-metabolizing enzymes in pig liver during subchronic dietary exposure to aflatoxin B1. Food Chem. Toxicol. 2007, 45, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Raysyan, A.; Eremin, S.A.; Beloglazova, N.V.; de Saeger, S.; Gravel, I.V. Immunochemical approaches for detection of aflatoxin B1 in herbal medicines. Phytochem. Anal. 2020, 31, 662–669. [Google Scholar] [CrossRef]
- Jedidi, I.; Cruz, A.; Jaen, M.T.G.; Said, S. Aflatoxins and ochratoxin A and their Aspergillus causal species in Tunisian cereals. Food Addit. Contam. B 2017, 10, 51–58. [Google Scholar] [CrossRef]
- Tang, Z. Hepatocellular Carcinoma-Cause, Treatment and Metastasis. World J. Gastroenterol. 2001, 7, 445. [Google Scholar] [CrossRef]
- Song, P.; Feng, X.; Zhang, K.; Song, T.; Ma, K.; Kokudo, N.; Dong, J.; Yao, L.; Tang, W. Screening for and surveillance of high-risk patients with HBV-related chronic liver disease: Promoting the early detection of hepatocellular carcinoma in China. BioSci. Trends 2013, 7, 1–6. [Google Scholar] [CrossRef]
- Martel, C.D.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015, 62, 1190–1200. [Google Scholar] [CrossRef]
- Ross, R.K.; Yu, M.C.; Henderson, B.E.; Yuan, J.M.; Qian, G.S.; Tu, J.T.; Gao, Y.T.; Wogan, G.N.; Groopman, J.D. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet 1992, 339, 943–946. [Google Scholar] [CrossRef]
- Qian, G.S.; Ross, R.K.; Yu, M.C.; Yuan, J.M.; Groopman, J.D. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People’s Republic of China. Cancer Epidemiol. Biomark. 1994, 3, 3–10. [Google Scholar]
- Wu, Y.L.; Peng, X.E.; Zhu, Y.B.; Yan, X.L.; Chen, W.N.; Lin, X. Hepatitis B virus X protein induces hepatic steatosis by enhancing the expression of liver fatty acid binding protein. J. Virol. 2016, 90, 1729–1740. [Google Scholar] [CrossRef]
- Niu, Y.; Wu, Z.; Shen, Q.; Song, J.; Luo, Q.; You, H.; Shi, G.; Qin, W. Hepatitis B virus X protein co-activates pregnane X receptor to induce the cytochrome P450 3A4 enzyme, a potential implication in hepatocarcinogenesis. Digest. Liver Dis. 2013, 45, 1041–1048. [Google Scholar] [CrossRef]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Liu, J.; Wang, L.Y.; Lu, S.N.; Lee, M.H.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of cirrhosis and hepatocellular carcinoma in chronic hepatitis B virus carriers. Int. J. Cancer 2017, 141, 711–720. [Google Scholar] [CrossRef]
- Corcuera, L.A.; Arbillaga, L.; Vettorazzi, A.; Azqueta, A.A. Lopez de Cerain, Ochratoxin A reduces aflatoxin B1 induced DNA damage detected by the comet assay in Hep G2 cells. Food Chem. Toxicol. 2011, 49, 2883–2889. [Google Scholar] [CrossRef]
- Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflatoxicosis in developing countries: A review of toxicology, exposure, potential health consequences, and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar] [CrossRef]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2010, 31, 71–82. [Google Scholar] [CrossRef]
- Eaton, D.L.; Groopman, J.D. The Toxicology of Aflatoxins: Human Health, Veterinary, and Agricultural Significance; Academic Press: Cambridge, MA, USA, 1994. [Google Scholar]
- Rotimi, O.A.; Rotimi, S.O.; Duru, C.U.; Ebebeinwe, O.J.; Abiodun, A.O.; Oyeniyi, B.O.; Faduyile, F.A. Acute aflatoxin B1—Induced hepatotoxicity alters gene expression and disrupts lipid and lipoprotein metabolism in rats. Toxicol. Rep. 2017, 4, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hu, B.; Shao, L.; Tian, Y.; Jin, T.; Jin, Y.; Ji, S.; Fan, X. Integrated analysis of transcriptomics and metabonomics profiles in aflatoxin B1-induced hepatotoxicity in rat. Food Chem. Toxicol. 2013, 55, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, Z.; Eeza, M.N.H.; Matysik, J.; Berry, J.P.; Alia, A. NMR-Based metabolic profiles of intact zebrafish embryos exposed to aflatoxin B1 recapitulates hepatotoxicity and supports possible neurotoxicity. Toxins 2019, 11, 258. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Hayton, S.; Maker, G.L.; Mullaney, I.; Trengove, R.D. Experimental design and reporting standards for metabolomics studies of mammalian cell lines. Cell Mol. Life Sci. 2017, 74, 4421–4441. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Tuluce, Y.; Isik, I. Evalution of toxicity of abcisic acid and gibberellic acid in rats: 50 days drinking water study. J. Enzym. Inhib. Med. Chem. 2008, 22, 219–226. [Google Scholar] [CrossRef]
- Mally, A.; Hard, G.C.; Dekant, W. Ochratoxin A as a potential etiologic factor in endemic nephropathy: Lessons from toxicity studies in rats. Food Chem. Toxicol. 2007, 45, 2254–2260. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Connelly, J.; Lindon, J.C.; Holmes, E. Metabonomics: A platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 2002, 1, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Slim, R.M.; Robertson, D.G.; Albassam, M.; Reily, M.D.; Robosky, L.; Dethloff, L.A. Effect of Dexamethasone on the Metabonomics Profile Associated with Phosphodiesterase Inhibitor-Induced Vascular Lesions in Rats. Toxicol. Appl. Pharm. 2002, 183, 108–116. [Google Scholar] [CrossRef]
- Liao, P.; Wei, L.; Zhang, X.; Li, X.; Wu, H.; Wu, Y.; Ni, J.; Pei, F. Metabolic profiling of serum from gadolinium chloride-treated rats by 1H NMR spectroscopy. Anal. Biochem. 2007, 364, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.G.; Reily, M.D.; Sigler, R.E.; Wells, D.F.; Paterson, D.A.; Braden, T.K. Metabonomics: Evaluation of Nuclear Magnetic Resonance (NMR) and Pattern Recognition Technology for Rapid in Vivo Screening of Liver and Kidney Toxicants. Toxicol. Sci. 2000, 57, 326–337. [Google Scholar] [CrossRef]
- Waterfield, C.J.; Turton, J.A.; Scales, M.D.; Timbrell, J.A. Investigations into the effects of various hepatotoxic compounds on urinary and liver taurine levels in rats. Arch. Toxicol. 1993, 67, 244–254. [Google Scholar] [CrossRef]
- Wu, H.; Feng, F. Untargeted metabolomic analysis using LC-TOF/MS and LC-MS/MS for revealing metabolic alterations linked to alcohol-induced hepatic steatosis in rat serum and plasma. RSC Adv. 2016, 6, 28279–28288. [Google Scholar] [CrossRef]
- Sasaki, H.; Akiyama, H.; Yoshida, Y.; Kondo, K.; Amakura, Y.; Kasahara, Y.; Maitan, T. Sugihiratake mushroom (Angel’s Wing Mushroom)-induced cryptogenic encephalopathy may involve vitamin D analogues. Biol. Pharm. Bull. 2006, 29, 2514–2518. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Zhang, Y.; Zheng, N.; Guo, L.; Song, X.; Zhao, S.; Wang, J. Biological system responses of dairy cows to aflatoxin B1 exposure revealed with metabolomic changes in multiple biofluids. Toxins 2019, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, S.; Fan, C.; Zheng, N.; Zhang, Y.; Li, S.; Wang, J. Metabolomic analysis of alterations in lipid oxidation, carbohydrate and amino acid metabolism in dairy goats caused by exposure to Aflotoxin B1. J. Dairy Res. 2017, 84, 401–406. [Google Scholar] [CrossRef]
- Bujak, R.; Struck-Lewicka, W.; Markuszewski, M.J.; Kaliszan, R. Metabolomics for laboratory diagnostics. J. Pharm. Biomed Anal. 2015, 113, 108–120. [Google Scholar] [CrossRef]
- Liu, W.; Wang, L.; Yang, X.; Zeng, H.; Zhang, R.; Pu, C.; Zheng, C.; Tan, Y.; Luo, Y.; Feng, X.; et al. Environmental microcystin exposure increases liver injury risk induced by hepatitis B virus combined with aflatoxin: A cross-sectional study in southwest china. Environ. Sci. Technol. 2017, 6, 6367–6378. [Google Scholar] [CrossRef]
- Fishbein, A.; Wang, W.; Yang, H. Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression. Proc. Natl. Acad. Sci. India B 2020, 117, 21576–21587. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xing, L.; Zhang, M.; Wang, J.; Zheng, N. The toxic effects of aflatoxin B1 and aflatoxin M1 on kidney through regulating L-proline and downstream apoptosis. Biomed Res. Int. 2018, 2018, 9074861. [Google Scholar] [CrossRef]
- Su, L.; Zhao, H.; Zhang, X.; Lou, Z.; Dong, X. UHPLC-Q-TOF-MS based serum metabonomics revealed the metabolic perturbations of ischemic stroke and the protective effect of RKIP in rat models. Mol. BioSyst. 2016, 12, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Pu, Z.J.; Kai, J.; Kang, A.; Tang, Y.P.; Shang, L.L.; Zhou, G.S.; Zhu, Z.H.; Shang, E.X.; Li, S.P.; et al. Comparative metabolomics analysis for the compatibility and incompatibility of kansui and licorice with different ratios by UHPLC-QTOF/MS and multivariate data analysis. J. Chromatogr. B 2017, 1057, 40–45. [Google Scholar] [CrossRef]
- Pedley, A.M.; Benkovic, S.J. A new View into the regulation of purine metabolism: The purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef]
- Wild, C.P.; Garner, R.C.; Ontesano, R.M.; Tursi, F. Aflatoxin B1 binding to plasma albumin and liver DNA upon chronic administration to rats. Carcinogenesis 1986, 7, 853–858. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, Y.; An, Y.; Tian, Y.; Wang, Y.; Tang, H. Systems responses of rats to aflatoxin B1 exposure revealed with metabonomic changes in multiple biological matrices. J. Proteome Res. 2011, 10, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Alseth, I.; Dalhus, B.; Bjørås, M. Inosine in DNA and RNA. Curr. Opin. Genet. Dev. 2014, 26, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, Z.H.; Stuart, B.; Wahle, B.; Bomann, W.; Ahr, H., Jr. Characteristic Expression Profiles Induced by Genotoxic Carcinogens in Rat Liver. Toxicol. Sci. 2004, 77, 19–34. [Google Scholar] [CrossRef]
- Lyer, R.S.; Coles, B.F.; Raney, K.D.; Thier, R.; Guengerich, F.P.; Harris, T.M. DNA Adduction by the Potent Carcinogen Aflatoxin B1:Mechanistic Studies. J. Am. Chem. Soc. 1994, 116, 1603–1609. [Google Scholar]
- Mclean, M.D.; Dutton, M.F. Cellular Interactions and metabolism of aflatoxin: An update. Pharmacol. Therapeut. 1995, 65, 163–192. [Google Scholar] [CrossRef]
- Siddiqui, A.; Ceppi, P. A non-proliferative role of pyrimidine metabolism in cancer. Mol. Metab. 2020, 35, 100962. [Google Scholar] [CrossRef]
- Roe, D.S.; Yang, B.Z.; Saban, C.V.; Struys, E.; Sweetman, L.; Roe, C.R. Differentiation of long-chain fatty acid oxidation disorders using alternative precursors and acylcarnitine profiling in fibroblasts. Mol. Genet. Metab. 2006, 87, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Okun, J.; Kölke, S.; Schulze, A.; Kohlmülle, D.; Olgemöller, K.; Lindne, M.; Hoffmann, G.; Wanders, R.; Mayatepek, E. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: Diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. BBA Mol. Cell Biol. L 2002, 1584, 91–98. [Google Scholar] [CrossRef]
- Bi, H.; Li, F.; Krausz, K.W.; Qu, A.; Johnson, C.H.; Gonzalez, F.J. Targeted metabolomics of serum acylcarnitines evaluates hepatoprotective effect of wuzhi tablet (schisandra sphenanthera extract) against acute acetaminophen toxicity. Evid. Based Complementary Altern. 2013, 2013, 985257. [Google Scholar]
- Bhattacharyya, S.; Yan, K.; Pence, L.; Simpson, P.; Gill, P.; Letzig, L.; Beger, R.; Sullivan, J.; Kearns, G.; Reed, M. Targeted liquid chromatography–mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark. Med. 2014, 8, 147–159. [Google Scholar] [CrossRef]
- Lu, X.; Tian, Y.; Zhao, Q.; Jin, T.; Xiao, S.; Fan, X. Integrated metabonomics analysis of the size-response relationship of silica nanoparticles-induced toxicity in mice. Nanotechnology 2011, 22, 055101. [Google Scholar] [CrossRef] [PubMed]
- Klein, J. Membrane breakdown in acute and chronic neurodegeneration: Focus on choline-containing phospholipids. J. Neural. Transm. 2000, 107, 1027–1063. [Google Scholar] [CrossRef]
- Gesing, A.; Karbownik, L.M. Protective effects of melatonin and N-acetylserotonin on aflatoxin B1-induced lipid peroxidation in rats. Cell Biochem. Funct. 2008, 26, 314–319. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Zhou, X.; Chen, X.; Huang, H.; Zhao, S.; Li, X.; Zhong, D. Metabolite profiling and identification of triptolide in rats. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2013, 939, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef]
- Li, H.; Li, S.; Yang, H.; Wang, Y.; Wang, J.; Zheng, N. L-proline alleviates kidney injury caused by AFB1 and AFM1 through regulating excessive apoptosis of kidney cells. Toxins 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ding, J. Sialylation is involved in cell fate decision during development, reprogramming and cancer progression. Protein Cell 2019, 10, 550–565. [Google Scholar] [CrossRef]
- Hanover, J.A. Glycan-dependent signaling: O-linked N-acetylglucosamine. FASEB J. 2001, 15, 1865–1876. [Google Scholar] [CrossRef]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lu, Y.; Wang, R.; Liu, S.; Hu, X.; Wang, H. Transport and metabolic profiling studies of amentoflavone in Caco-2 cells by UHPLC-ESI-MS/MS and UHPLC-ESI-Q-TOF-MS/MS. J. Pharm. Biomed. Anal. 2020, 189, 113441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Wu, H.; Wu, H.; Liu, X.; Zhou, A. Metabolic profiling of copper-laden hepatolenticular degeneration model rats and the interventional effects of Gandou decoction using UPLC-Q-TOF/MS. J. Pharmaceut. Biomed. 2019, 164, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Number | Total | Hits | p-Value | Impact | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 16/DMSO | 32/DMSO | 32/16 | 16/DMSO | 32/DMSO | 32/16 | 16/DMSO | 32/DMSO | 32/16 | ||
| 1 | 65 | 13 | 12 | 9 | 2.26 × 10−5 | 1.05 × 10−3 | 2.38 × 10−3 | 0.24 | 0.16 | 0.15 |
| 2 | 28 | 6 | 6 | 3 | 3.05 × 10−3 | 9.69 × 10−3 | / | 0.49 | 0.56 | 0.27 |
| 3 | 39 | 7 | 7 | 5 | 3.99 × 10−3 | 1.42 × 10−2 | 3.15 × 10−2 | 0.20 | 0.26 | 0.24 |
| 4 | 14 | 3 | / | 2 | 3.62 × 10−2 | / | / | 0.23 | / | 0.23 |
| 5 | 6 | 2 | 2 | 1 | 3.78 × 10−2 | / | / | 0.50 | 0.50 | 0.50 |
| 6 | 36 | 5 | 6 | 3 | 4.15 × 10−2 | 3.21 × 10−2 | / | 0.24 | 0.27 | 0.22 |
| 7 | 19 | / | 4 | 3 | / | 3.60 × 10−2 | / | / | 0.04 | 0.18 |
| 8 | 9 | / | 2 | 2 | / | / | / | / | 0.57 | 0.57 |
| 9 | 38 | / | 4 | 4 | / | / | / | / | 0.26 | 0.10 |
| 10 | 39 | / | / | 3 | / | / | / | / | / | 0.12 |
| 11 | 28 | 4 | / | 2 | / | / | / | 0.34 | / | 0.28 |
| 12 | 33 | / | 3 | 2 | / | / | / | / | 0.23 | 0.23 |
| 13 | 36 | / | 2 | 2 | / | / | / | / | 0.31 | 0.31 |
| 14 | 15 | 2 | 2 | / | / | / | / | 0.14 | 0.14 | / |
| 15 | 4 | 1 | 1 | / | / | / | / | 0.20 | 0.25 | / |
| 16 | 10 | 1 | 1 | / | / | / | / | 0.36 | 0.36 | / |
| 17 | 30 | 1 | 1 | / | / | / | / | 0.13 | 0.13 | / |
| 18 | 41 | 1 | 1 | / | / | / | / | 0.14 | 0.14 | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Yang, X.; Liu, F.; Wang, X.; Zhang, X.; He, K.; Wang, H. Comprehensive Metabolomic Analysis Reveals Dynamic Metabolic Reprogramming in Hep3B Cells with Aflatoxin B1 Exposure. Toxins 2021, 13, 384. https://doi.org/10.3390/toxins13060384
Wang S, Yang X, Liu F, Wang X, Zhang X, He K, Wang H. Comprehensive Metabolomic Analysis Reveals Dynamic Metabolic Reprogramming in Hep3B Cells with Aflatoxin B1 Exposure. Toxins. 2021; 13(6):384. https://doi.org/10.3390/toxins13060384
Chicago/Turabian StyleWang, Shufeng, Xin Yang, Feng Liu, Xinzheng Wang, Xuemin Zhang, Kun He, and Hongxia Wang. 2021. "Comprehensive Metabolomic Analysis Reveals Dynamic Metabolic Reprogramming in Hep3B Cells with Aflatoxin B1 Exposure" Toxins 13, no. 6: 384. https://doi.org/10.3390/toxins13060384
APA StyleWang, S., Yang, X., Liu, F., Wang, X., Zhang, X., He, K., & Wang, H. (2021). Comprehensive Metabolomic Analysis Reveals Dynamic Metabolic Reprogramming in Hep3B Cells with Aflatoxin B1 Exposure. Toxins, 13(6), 384. https://doi.org/10.3390/toxins13060384