Molecular and Structural Basis of Cross-Reactivity in M. tuberculosis Toxin–Antitoxin Systems

 , ,
, ,
Abstract
1. Introduction
2. Results
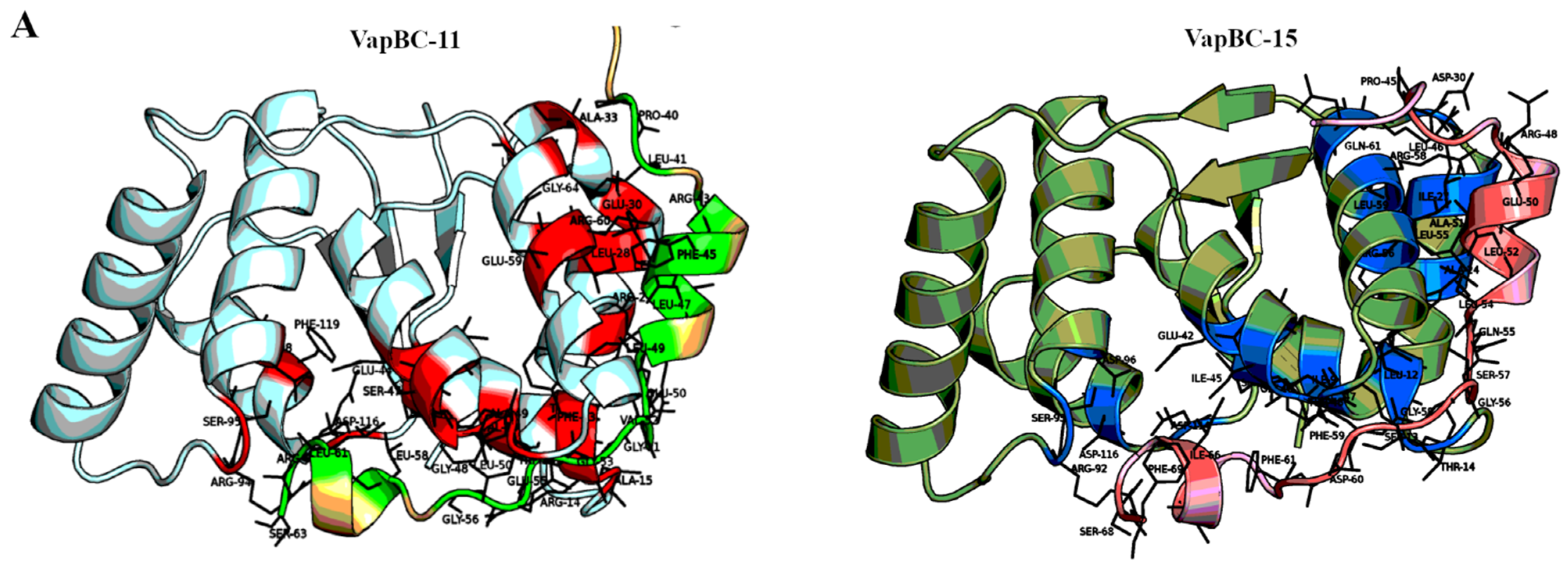
2.1. Comparisons of TA Interfaces Predict Cases for Potential Cross-Talk and Reveal Reasons for Insulation in Others
2.2. Templates Could Be Identified for Toxins at a Higher Confidence than Antitoxins
2.3. High Confidence Complexes Could Be Generated for Select TA Systems
2.4. Assessment of the Modelled Complexes Reveals Strengths and Limitations of Homology Modelling of TA Systems
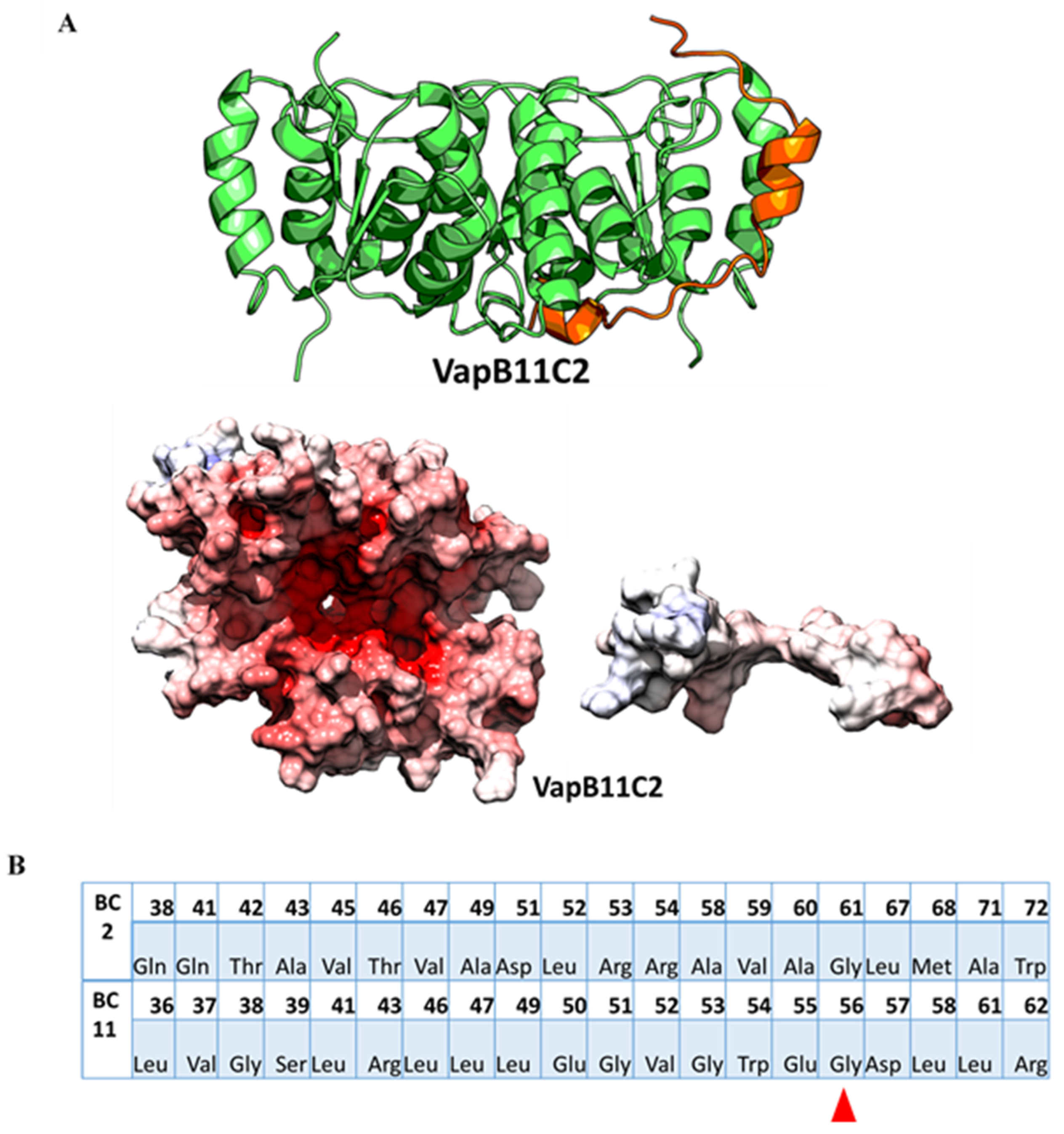
2.5. Use of High Confidence Modelled Complexes to Explore Cross-Reactivity
2.6. In-Silico Point Mutation of Antitoxin Residues Is Predicted to Relax the Specificity
2.7. Modelled Non-Cognate TA Pairs that Fail to Show Cross-Talk in Experiments Differ in Their Interfaces
2.8. MazEF Systems from M. tuberculosis Show Weak Signals for Cross-Reactivity
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Comparative Modelling of Toxins, Antitoxins and TA Complexes
5.2. Assessment of Toxin–Antitoxin Interfaces
- Surface complementarity was checked using Sc statistics, which measures the geometric surface complementarity of protein–protein interfaces [53]. Sc depends on the relative shape of the surfaces with respect to each other and on the extent to which the interaction brings individual elements of opposing surfaces into proximity. The score ranges between 0 and 1 and the threshold is generally decided based on the shape complementarity between antigen–antibody interfaces, where the weakest shape complementarity interface is reported with Sc values between 0.64 and 0.69.
- Electrostatic surface complementarity was assessed by calculating the electrostatic potential of the proteins. Hydrogens were added to the individual proteins. Charges were assigned to the residues using PDB2PQR [54] plugin and electrostatics calculation was performed using APBS [55] plugin in Chimera 1.13.1. The molecular surface was then color based on electrostatic potential and scaled between ±10 kBT and manually inspected.
- Interaction energy between toxin and antitoxin was calculated using AnalyseComplex module from FoldX package [28]. FoldX force-field is empirical in nature with terms for de-solvation energies, coulombic interactions, van der Waal’s forces, hydrogen bonding, entropic changes, and others. All the structures were energy minimized with GROMACS v5.1 using CHARMM27 force-field and steepest descent method for either 50,000 steps or till convergence. A dodecahedron box with TIP3P water molecules was defined around the protein and the system was neutralized by adding counter ions prior to minimization. The structures were further repaired for any distorted geometry using RepairPDB module from FoldX prior to energy calculations. The complex structures were minimized iteratively till no further improvement in the energy values.
5.3. Using Structures to Explore Cross-Reactivity between Non-Cognate Toxins and Antitoxins
5.4. Prediction of Hotspot Residues at the Interface and In-Silico Mutations Using FoldX
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayes, F.; Van Melderen, L. Toxins-Antitoxins: Diversity, Evolution and Function. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 386–408. [Google Scholar] [CrossRef]
- Kedzierska, B.; Hayes, F. Emerging Roles of Toxin-Antitoxin Modules in Bacterial Pathogenesis. Molecules 2016, 21, 790. [Google Scholar] [CrossRef]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin-Antitoxin Systems: Their Role in Persistence, Biofilm Formation, and Pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-del Portillo, F. Toxin-Antitoxins and Bacterial Virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef]
- Coray, D.S.; Wheeler, N.E.; Heinemann, J.A.; Gardner, P.P. Why so Narrow: Distribution of Anti-Sense Regulated, Type I Toxin-Antitoxin Systems Compared with Type II and Type III Systems. RNA Biol. 2017, 14, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Drze, P.; Van Melderen, L. Diversity of Bacterial Type II Toxin-Antitoxin Systems: A Comprehensive Search and Functional Analysis of Novel Families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; De Bast, M.S. Bacterial Toxin-Antitoxin Systems: More than Selfish Entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef] [PubMed]
- Goeders, N.; Van Melderen, L. Toxin-Antitoxin Systems as Multilevel Interaction Systems. Toxins 2013, 6, 304–324. [Google Scholar] [CrossRef]
- Park, J.H.; Yoshizumi, S.; Yamaguchi, Y.; Wu, K.P.; Inouye, M. ACA-Specific RNA Sequence Recognition Is Acquired via the Loop 2 Region of MazF MRNA Interferase. Proteins Struct. Funct. Bioinform. 2013, 81, 874–883. [Google Scholar] [CrossRef]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive Functional Analysis of Mycobacterium tuberculosis Toxin-Antitoxin Systems: Implications for Pathogenesis, Stress Responses, and Evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef]
- Walling, L.R.; Butler, J.S. Structural Determinants for Antitoxin Identity and Insulation of Cross Talk between Homologous Toxin-Antitoxin Systems. J. Bacteriol. 2016, 198, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gao, C.; Wang, Y.; Zhang, H.; He, Z.G. Characterization of the Interaction and Cross-Regulation of Three Mycobacterium tuberculosis RelBE Modules. PLoS ONE 2010, 5, e10672. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Sharp, J.D.; Kobayashi, H.; Woychik, N.A.; Inouye, M. Noncognate Mycobacterium tuberculosis Toxin-Antitoxins Can Physically and Functionally Interact. J. Biol. Chem. 2010, 285, 39732–39738. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.V.; Dawson, C.C.; Crew, R.; England, K.; Slayden, R.A. MazF6 Toxin of Mycobacterium tuberculosis Demonstrates Antitoxin Specificity and Is Coupled to Regulation of Cell Growth by a Soj-like Protein. BMC Microbiol. 2013, 13, 240. [Google Scholar] [CrossRef] [PubMed]
- Riffaud, C.; Pinel-Marie, M.L.; Pascreau, G.; Felden, B. Functionality and Cross-Regulation of the Four SprG/SprF Type I Toxin-Antitoxin Systems in Staphylococcus aureus. Nucleic Acids Res. 2019, 47, 1740–1758. [Google Scholar] [CrossRef]
- Chen, R.; Tu, J.; Tan, Y.; Cai, X.; Yang, C.; Deng, X.; Su, B.; Ma, S.; Liu, X.; Ma, P.; et al. Structural and Biochemical Characterization of the Cognate and Heterologous Interactions of the MazEF-Mt9 TA System. ACS Infect. Dis. 2019, 5, 1306–1316. [Google Scholar] [CrossRef]
- Wei, Y.; Li, Y.; Yang, F.; Wu, Q.; Liu, D.; Li, X.; Hua, H.; Liu, X.; Wang, Y.; Zheng, K.; et al. Physical and Functional Interplay between MazF 1Bif and Its Noncognate Antitoxins from Bifidobacterium longum. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Smith, A.B.; López-Villarejo, J.; Diago-Navarro, E.; Mitchenall, L.A.; Barendregt, A.; Heck, A.J.; Lemonnier, M.; Maxwell, A.; Díaz-Orejas, R. A Common Origin for the Bacterial Toxin-Antitoxin Systems ParD and Ccd, Suggested by Analyses of Toxin/Target and Toxin/Antitoxin Interactions. PLoS ONE 2012, 7, e46499. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Struct. Bioinform. 2005, 181–198. [Google Scholar] [CrossRef]
- Tandon, H.; Sharma, A.; Wadhwa, S.; Varadarajan, R.; Singh, R.; Srinivasan, N.; Sandhya, S. Bioinformatic and Mutational Studies of Related Toxin–Antitoxin Pairs in Mycobacterium tuberculosis Predict and Identify Key Functional Residues. J. Biol. Chem. 2019, 294, 9048–9063. [Google Scholar] [CrossRef]
- Min, A.B.; Miallau, L.; Sawaya, M.R.; Habel, J.; Cascio, D.; Eisenberg, D. The Crystal Structure of the Rv0301-Rv0300 VapBC-3 Toxin-Antitoxin Complex from M. tuberculosis Reveals a Mg2+ Ion in the Active Site and a Putative RNA-Binding Site. Protein Sci. 2012, 21, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Tiwari, P.; Agarwal, S.; Kaundal, S.; Kidwai, S.; Singh, R.; Thakur, K.G. Structural, Functional and Biological Insights into the Role of Mycobacterium tuberculosis VapBC11 Toxin–Antitoxin System: Targeting a TRNase to Tackle Mycobacterial Adaptation. Nucleic Acids Res. 2018, 46, 11639–11655. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Pogenberg, V.; Subhramanyam, U.K.T.; Wilmanns, M.; Gourinath, S.; Srinivasan, A. Crystal Structure of the VapBc-15 Complex from Mycobacterium tuberculosis Reveals a Two-Metal Ion Dependent Pin-Domain Ribonuclease and a Variable Mode of Toxin-Antitoxin Assembly. J. Struct. Biol. 2014, 188, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Jardim, P.; da Silva Santos, I.C.; Barbosa, J.A.R.G.; de Freitas, S.M.; Valadares, N.F. Crystal Structure of VapC21 from Mycobacterium tuberculosis at 1.31 Å Resolution. Biochem. Biophys. Res. Commun. 2016, 478, 1370–1375. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Wallner, B.; Elofsson, A. Can Correct Protein Models Be Identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Pugalenthi, G.; Shameer, K.; Srinivasan, N.; Sowdhamini, R. HARMONY: A Server for the Assessment of Protein Structures. Nucleic Acids Res. 2006, 34, W231–W234. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33 (Suppl. 2), W382–W388. [Google Scholar] [CrossRef]
- Yen, T.J.; Brennan, R.G. Crystal Structure of M. tuberculosis MazF-Mt3 (Rv1991c) in Complex with RNA. Ph.D. Thesis, Rutgers University-School of Graduate Studies, Rutgers, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Yen, T.J.; Brennan, R.G. Structure of M. tuberculosis MazF-Mt1 (Rv2801c) in Complex with RNA. ACS Infect. Dis. 2017. [Google Scholar] [CrossRef]
- Hoffer, E.D.; Miles, S.J.; Dunham, C.M. The Structure and Function of Mycobacterium tuberculosis MazF-Mt6 Toxin Provide Insights into Conserved Features of MazF Endonucleases. J. Biol. Chem. 2017, 292, 7718–7726. [Google Scholar] [CrossRef]
- Zorzini, V.; Mernik, A.; Lah, J.; Sterckx, Y.G.J.; De Jonge, N.; Garcia-Pino, A.; De Greve, H.; Verse, W.; Loris, R. Substrate Recognition and Activity Regulation of the Escherichia Coli MRNA Endonuclease MazF. J. Biol. Chem. 2016, 291, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal Structure of the MazE/MazF Complex: Molecular Bases of Antidote-Toxin Recognition. Mol. Cell 2003, 11, 875–884. [Google Scholar] [CrossRef]
- Miallau, L.; Faller, M.; Chiang, J.; Arbing, M.; Guo, F.; Cascio, D.; Eisenberg, D. Structure and Proposed Activity of a Member of the VapBC Family of Toxin-Antitoxin Systems. J. Biol. Chem. 2009, 284, 276–283. [Google Scholar] [CrossRef]
- Jin, G.; Pavelka, M.S.; Butler, J.S. Structure-Function Analysis of VapB4 Antitoxin Identifies Critical Features of a Minimal VapC4 Toxin-Binding Module. J. Bacteriol. 2015, 197, 1197–1207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thorn, K.S.; Bogan, A.A. ASEdb: A Database of Alanine Mutations and Their Effects on the Free Energy of Binding in Protein Interactions. Bioinformatics 2001, 17, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, S.; Sukhwal, A.; Sowdhamini, R.; Srinivasan, N. Specificity and Stability of Transient Protein–Protein Interactions. Curr. Opin. Struct. Biol. 2017, 44, 77–86. [Google Scholar] [CrossRef]
- Sala, A.; Bordes, P.; Genevaux, P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002–1020. [Google Scholar] [CrossRef]
- Chen, R.; Tu, J.; Liu, Z.; Meng, F.; Ma, P.; Ding, Z.; Yang, C.; Chen, L.; Deng, X.; Xie, W. Structure of the MazF-Mt9 Toxin, a TRNA-Specific Endonuclease from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 2017, 486, 804–810. [Google Scholar] [CrossRef]
- Agarwal, S.; Tiwari, P.; Deep, A.; Kidwai, S.; Gupta, S.; Thakur, K.G.; Singh, R. System-Wide Analysis Unravels the Differential Regulation and in Vivo Essentiality of Virulence-Associated Proteins B and C Toxin-Antitoxin Systems of Mycobacterium tuberculosis. J. Infect. Dis. 2018, 217, 1809–1820. [Google Scholar] [CrossRef]
- Gupta, A.; Venkataraman, B.; Vasudevan, M.; Gopinath Bankar, K. Co-Expression Network Analysis of Toxin-Antitoxin Loci in Mycobacterium tuberculosis Reveals Key Modulators of Cellular Stress. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Frampton, R.; Aggio, R.B.M.; Villas-Bôas, S.G.; Arcus, V.L.; Cook, G.M. Toxin-Antitoxin Systems of Mycobacterium smegmatis Are Essential for Cell Survival. J. Biol. Chem. 2012, 287, 5340–5356. [Google Scholar] [CrossRef]
- Aloy, P.; Ceulemans, H.; Stark, A.; Russell, R.B. The Relationship between Sequence and Interaction Divergence in Proteins. J. Mol. Biol. 2003, 332, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Yazhini, A.; Srinivasan, N. How Good Are Comparative Models in the Understanding of Protein Dynamics? Proteins Struct. Funct. Bioinform. 2020, 88, 874–888. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Söding, J. Fast and Accurate Automatic Structure Prediction with HHpred. Proteins Struct. Funct. Bioinform. 2009, 77 (Suppl. 9), 128–132. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma. 2016, 2016, 5.6.1–5.6.37. [Google Scholar]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L. Improved Prediction of Protein Side-Chain Conformations with SCWRL4. Proteins Struct. Funct. Bioinform. 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Baspinar, A.; Cukuroglu, E.; Nussinov, R.; Keskin, O.; Gursoy, A. PRISM: A Web Server and Repository for Prediction of Protein-Protein Interactions and Modeling Their 3D Complexes. Nucleic Acids Res. 2014, 42(W1), W285–W289. [Google Scholar] [CrossRef]
- Kundrotas, P.J.; Zhu, Z.; Janin, J.; Vakser, I.A. Templates Are Available to Model Nearly All Complexes of Structurally Characterized Proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 9438–9441. [Google Scholar] [CrossRef] [PubMed]
- Rajagopala, S.V.; Sikorski, P.; Kumar, A.; Mosca, R.; Vlasblom, J.; Arnold, R.; Franca-Koh, J.; Pakala, S.B.; Phanse, S.; Ceol, A.; et al. The Binary Protein-Protein Interaction Landscape of Escherichia coli. Nat. Biotechnol. 2014, 32, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Colman, P. Shape Complementarity at Protein–Protein Interfaces. J. Mol. Biol. 1993, 234, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J.; Rost, B. IAlign: A Method for the Structural Comparison of Protein-Protein Interfaces. Bioinformatics 2011, 27, 2259–2265. [Google Scholar] [CrossRef]
- Christen, M.; Hünenberger, P.H.; Bakowies, D.; Baron, R.; Bürgi, R.; Geerke, D.P.; Heinz, T.N.; Kastenholz, M.A.; Kräutler, V.; Oostenbrink, C.; et al. The GROMOS Software for Biomolecular Simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex Structures to be Compared (Vap) | IS-Score | z-Score | % Seq. Identity | No. of Aligned Contacts | RMSD (Ǻ) | p-Value |
|---|---|---|---|---|---|---|
| BC2–BC11 | 0.25 | 5 | 10 | 24 | 3.11 | 3.2 × 10−3 |
| BC2–BC15 | 0.24 | 5 | 5 | 23 | 3.27 | 3 × 10−3 |
| BC11–BC15 | 0.52 | 23 | 39 | 48 | 1.98 | 4.2 × 10−11 |
| BC4–BC5 | 0.81 | 45 | 40 | 70 | 0.89 | 2.5 × 10−19 |
| Complex Structure | IS-Score | z-Score | % Seq. Identity | No. of Aligned Contacts | RMSD (Å) | p-Value | Shape Complementarity for Non-Cognate Pair (Sc) |
|---|---|---|---|---|---|---|---|
| BC15–B11C15 | 0.57 | 24 | 70 | 40 | 1.62 | 3.8 × 10−11 | 0.67 |
| BC11–B15C11 | 0.54 | 23 | 76 | 38 | 1.46 | 8.3 × 10−11 | 0.60 |
| BC4–B5C4 | 0.81 | 49 | 75 | 79 | 0.25 | 2.2 × 10−21 | 0.67 |
| BC5–B4C5 | 0.83 | 49 | 83 | 80 | 0.23 | 2.0 × 10−21 | 0.65 |
| TA System | Sc Statistics | Interaction Energy (Kcal/mol) |
|---|---|---|
| VapBC3 | 0.59 | - |
| VapBC4 | 0.64 | −35.04 |
| VapBC21 | 0.64 | −15.83 |
| MazEF3 | 0.68 & 0.445 | −17.2 |
| MazEF6 | 0.67 & 0.67 | −22.2 |
| MazEF9 | 0.62 & 0.65 | −16.2 |
| Complex Structure | IS-Score | z-Score | % Seq. Identity | RMSD (Å) | p-Value | Shape Complementarity (Sc) |
|---|---|---|---|---|---|---|
| BC2–B11C2 | 0.3 | 9.5 | No residue from antitoxin | 2.9 | 7 × 10−5 | 0.60 |
| BC11–B2C11 | 0.25 | 5.7 | No residue from antitoxin | 3.18 | 3.3 × 10−3 | 0.59 |
| Complex Structure to be Compared (Maz) | IS-Score | z-Score | % Seq. Identity | No. of Aligned Contacts | RMSD (Å) | p-Value |
|---|---|---|---|---|---|---|
| EF4-EF6 | 0.16 | 1.5 | 35 | 7 | 2.38 | 1.9 × 10−1 |
| EF4-EF3 | 0.45 | 16.3 | 30 | 18 | 2.39 | 7.5 × 10−8 |
| EF4-EF7 | 0.12 | −0.4 | 10 | 3 | 1.78 | 0.8 × 10−1 |
| EF4-EF9 | 0.16 | 0.6 | 0 | 8 | 2.75 | 3.9 × 10−1 |
| EF3-EF6 | 0.15 | 0.84 | 25 | 4 | 2.32 | 3.4 × 10−1 |
| EF3-EF7 | 0.13 | 0.34 | 11 | 4 | 3.52 | 5.0 × 10−1 |
| EF6-EF7 | 0.20 | 3.75 | 4 | 12 | 3.00 | 2.3 × 10−2 |
| EF6-EF9 | 0.38 | 15.3 | 7 | 20 | 1.56 | 2.4 × 10−7 |
| EF3-EF9 | 0.14 | 0.12 | 0 | 8 | 3.81 | 5.8 × 10−1 |
| EF7-EF9 | 0.18 | 2.67 | 20 | 13 | 3.13 | 6.6 × 10−2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tandon, H.; Melarkode Vattekatte, A.; Srinivasan, N.; Sandhya, S. Molecular and Structural Basis of Cross-Reactivity in M. tuberculosis Toxin–Antitoxin Systems. Toxins 2020, 12, 481. https://doi.org/10.3390/toxins12080481
Tandon H, Melarkode Vattekatte A, Srinivasan N, Sandhya S. Molecular and Structural Basis of Cross-Reactivity in M. tuberculosis Toxin–Antitoxin Systems. Toxins. 2020; 12(8):481. https://doi.org/10.3390/toxins12080481
Chicago/Turabian StyleTandon, Himani, Akhila Melarkode Vattekatte, Narayanaswamy Srinivasan, and Sankaran Sandhya. 2020. "Molecular and Structural Basis of Cross-Reactivity in M. tuberculosis Toxin–Antitoxin Systems" Toxins 12, no. 8: 481. https://doi.org/10.3390/toxins12080481
APA StyleTandon, H., Melarkode Vattekatte, A., Srinivasan, N., & Sandhya, S. (2020). Molecular and Structural Basis of Cross-Reactivity in M. tuberculosis Toxin–Antitoxin Systems. Toxins, 12(8), 481. https://doi.org/10.3390/toxins12080481