Enzyme Immunoassay for Measuring Aflatoxin B1 in Legal Cannabis

 ,
, 
 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Enzyme Immunoassay Adaptation to AFB1 Detection in Cannabis Products
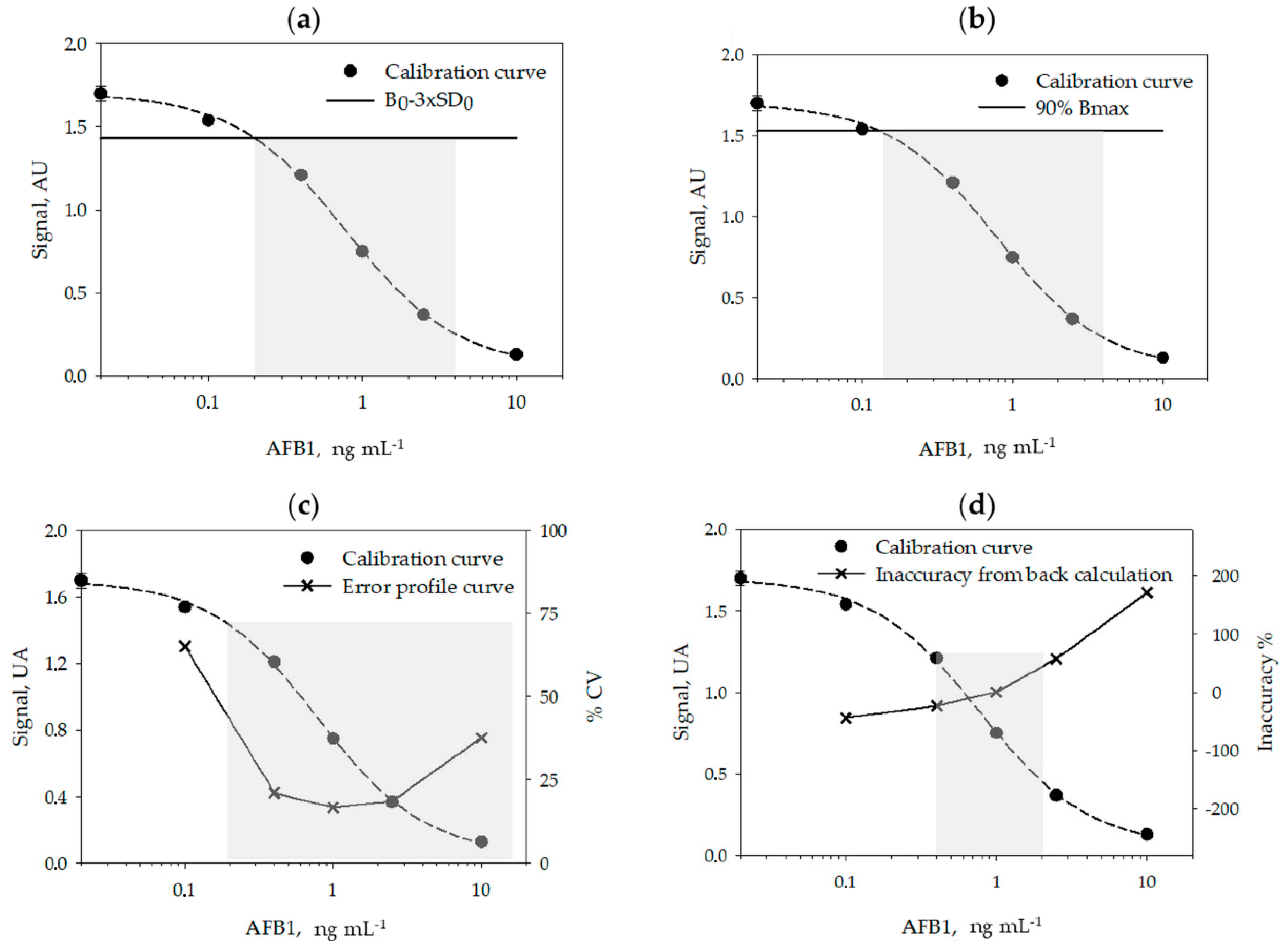
2.2. Analytical Figures of Merits of the Enzyme Immunoassay
2.3. Measuring AFB1 in Cannabis Products by the Enzyme Immunoassay
2.4. Liquid Chromatography Tandem Mass Spectrometry Determination of AFB1 in Cannabis Products
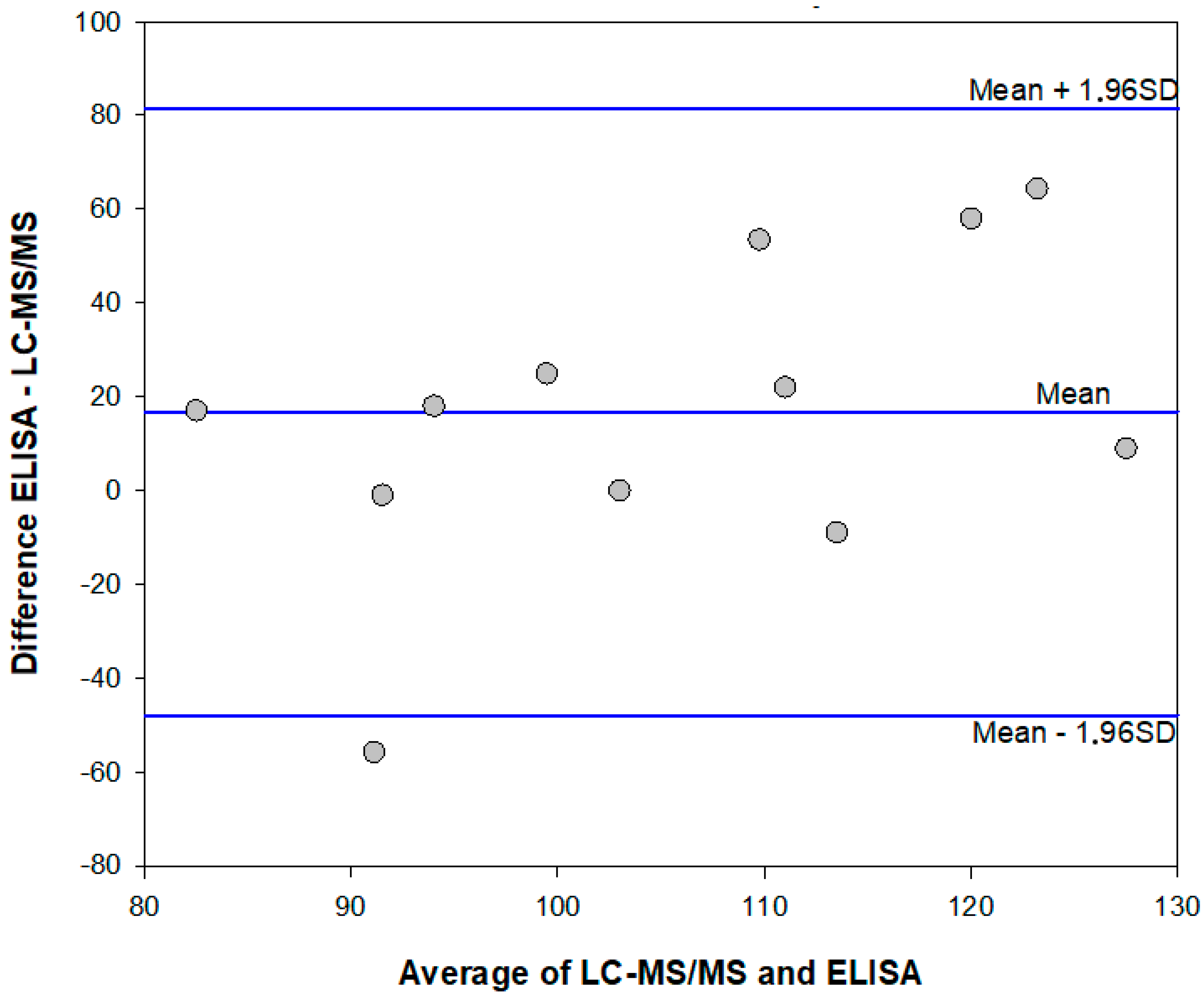
2.5. Method Comparison: Enzyme Immunoassay and HPLC-MS/MS
3. Discussion
4. Materials and Methods
4.1. Reagents and Apparatus
4.2. Competitive Direct ELISA
4.3. Cross-Reactivity Study
4.4. Samples and Sample Preparation
4.5. Liquid Chromatography Coupled to Tandem Mass Spectrometry Detection of AFB1
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- FDA. Regulation of Cannabis and Cannabis-Derived Products: Questions and Answer. Available online: https://www.fda.gov/news-events/public-health-focus/fda-regulation-cannabis-and-cannabis-derived-products-questions-and-answers (accessed on 10 July 2019).
- Mead, A. Legal and Regulatory Issues Governing Cannabis and Cannabis-Derived Products in the United States. Front. Plant Sci. 2019, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Hartsel, J.A.; Eades, J.; Hickory, B.; Makriyannis, A. Chapter 53—Cannabis Sativa and Hemp in Nutraceuticals Efficacy, Safety and Toxicity; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 735–754. [Google Scholar]
- FDA. Regulation of Cannabis and Cannabis-Derived Products, Including Cannabidiol (CBD). Available online: https://www.fda.gov/news-events/public-health-focus/fda-regulation-cannabis-and-cannabis-derived-products-including-cannabidiol-cbd (accessed on 1 March 2020).
- Management of Substance Abuse—Cannabis. Available online: https://www.who.int/substance_abuse/facts/cannabis/en/ (accessed on 20 April 2020).
- Bridgeman, M.B.; Abazia, D.T. Medicinal Cannabis: History, Pharmacology, and Implications for the Acute Care Setting. Pharm. Ther. 2017, 42, 180–188. [Google Scholar]
- Medical Use of Cannabis and Cannabinoids—Questions and Answers for Policymaking. Available online: www.emcdda.europa.eu›system›files›publications (accessed on 4 November 2019).
- Corroon, J.; Kight, R. Regulatory Status of Cannabidiol in the United States: A Perspective. Cannabis Cannabinoid Res. 2018, 3, 190–194. [Google Scholar] [CrossRef] [PubMed]
- EU. Regulation 1307/2013. Off. J. Eur. Union 2013, L347, 608–670. [Google Scholar]
- Dryburgh, L.M.; Bolan, N.S.; Grof, C.P.; Galettis, P.; Schneider, J.; Lucas, C.J.; Martin, J.H. Cannabis contaminants: Sources, distribution, human toxicity and pharmacologic effects. Br. J. Clin. Pharmacol. 2018, 84, 2468–2476. [Google Scholar] [CrossRef]
- Zerihun, A.; Chandravanshi, A.S.; Debebe, A.; Mehari, B. Levels of selected metals in leaves of Cannabis sativa L. cultivated in Ethiopia. Springerplus 2015, 4, e359. [Google Scholar] [CrossRef]
- Llewellyn, G.C.; O’Rear, C.E. Examination of fungal growth and aflatoxin production on marihuana. Mycopathologia 1977, 62, 109–112. [Google Scholar] [CrossRef]
- Wilcox, J.; Pazdanska, M.; Milligan, C.; Chan, D.; MacDonald, S.J.; Donnelly, C. Analysis of Aflatoxins and Ochratoxin A in Cannabis and Cannabis Products by LC-Fluorescence Detection Using Cleanup with Either Multiantibody Immunoaffinity Columns or an Automated System with In-Line Reusable Immunoaffinity Cartridges. J. AOAC Int. 2019. [Google Scholar] [CrossRef]
- McKernan, K.; Spangler, J.; Zhang, L.; Tadigotla, V.; Helbert, Y.; Foss, T.; Smith, D. Cannabis microbiome sequencing reveals several mycotoxic fungi native to dispensary grade Cannabis flowers. F1000Research 2015, 4, 1422. [Google Scholar] [CrossRef]
- EFSA. Aflatoxins in Food. Available online: https://www.efsa.europa.eu/en/topics/topic/aflatoxins-food (accessed on 10 July 2019).
- Marchese, S.; Polo, A.; Ariano, A.; Velotto, S.; Costantini, S.; Severino, L. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins 2018, 10, 214. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer Monograph on the Evaluation of Carcinogenic Risks in Humans. Some Traditional Herbal Medicines, Some Mycotoxins Naphthalene and Styrene; IARC: Lyon, France, 2002; Volume 82, pp. 171–274. [Google Scholar]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, L.; Di Nardo, F.; Giovannoli, C.; Passini, C.; Baggiani, C. Enzyme immunoassay for monitoring aflatoxins in eggs. Food Control 2015, 57, 115–121. [Google Scholar] [CrossRef]
- Ventura, M.; Gómez, A.; Anaya, I.; Díaz, I.; Broto, F.; Agut, M.; Comellas, L. Determination of aflatoxins B1, G1, B2 and G2 in medicinal herbs by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2004, 1048, 25–29. [Google Scholar]
- Arranz, I.; Sizoo, E.; van Egmond, H.; Kroeger, K.; Legarda, T.M.; Burdaspal, P.; Reif, K.; Stroka, J. Determination of aflatoxin B1 in medical herbs: Interlaboratory study. J. AOAC Int. 2006, 89, 595–605. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, S.; Ge, D.; Zhu, N.; Wang, K.; Zhu, G.; Xu, W.; Xu, H. An ultrasensitive competitive immunosensor using silica nanoparticles as an enzyme carrier for simultaneous impedimetric detection of tetrabromobisphenol A bis(2-hydroxyethyl) ether and tetrabromobisphenol A mono(hydroxyethyl) ether. Biosens. Bioelectron. 2018, 105, 77–80. [Google Scholar] [CrossRef]
- Reimer, G.J.; Gee, S.J.; Hammock, B.D. Comparison of a Time-Resolved Fluorescence Immunoassay and an Enzyme-Linked Immunosorbent Assay for the Analysis of Atrazine in Water. J. Agric. Food Chem. 1998, 46, 3353–3358. [Google Scholar] [CrossRef]
- Sasaki, D.; Mitchell, R.A. How to Obtain Reproducible Quantitative ELISA Results; Oxford Biomedical Research Inc.: Oxford, UK, 2001. [Google Scholar]
- Saeed, A.F.U.H.; Ling, S.; Yuan, J.; Wang, S. The Preparation and Identification of a Monoclonal Antibody against Domoic Acid and Establishment of Detection by Indirect Competitive ELISA. Toxins 2017, 9, 250. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, X.; Wen, K.; Li, C.; Mari, G.M.; Jiang, H.; Shi, W.; Shen, J.; Wang, Z. Multiplex Lateral Flow Immunoassays Based on Amorphous Carbon Nanoparticles for Detecting Three Fusarium Mycotoxins in Maize. J. Agric. Food Chem. 2017, 65, 8063–8071. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, N.; Zou, Y.; Zhao, Z.; Wu, W.; Liang, G.; Han, Z.; Meng, H. A novel and sensitive chemiluminescence immunoassay based on AuNCs@pepsin@luminol for simultaneous detection of tetrabromobisphenol A bis(2-hydroxyethyl) ether and tetrabromobisphenol A mono(hydroxyethyl) ether. Anal. Chim. Acta 2018, 1035, 168–174. [Google Scholar] [CrossRef]
- Quinn, C.P.; Semenova, V.A.; Elie, C.M.; Romero-Steiner, S.; Greene, C.; Li, H.; Stamey, K.; Steward-Clark, E.; Schmidt, D.S.; Mothershed, E.; et al. Specific, Sensitive, and Quantitative Enzyme-Linked Immunosorbent Assay for Human Immunoglobulin G Antibodies to Anthrax Toxin Protective Antigen. Emerg. Infect. Dis. 2002, 8, 1103–1110. [Google Scholar] [CrossRef]
- Dunn, J.; Wild, D. Calibration curve fitting. In The Immunoassay Handbook, 4th ed.; Wild, D., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; pp. 323–336. [Google Scholar]
- Zheng, R.; Xu, H.; Wang, W.; Zhan, R.; Chen, W. Simultaneous determination of aflatoxin B(1), B(2), G(1), G(2), ochratoxin A, and sterigmatocystin in traditional Chinese medicines by LC-MS-MS. Anal. Bioanal. Chem. 2014, 406, 3031–3039. [Google Scholar] [CrossRef] [PubMed]
- Narváez, A.; Rodríguez-Carrasco, Y.; Castaldo, L.; Izzo, L.; Ritieni, A. Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals. Toxins 2020, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Peckham, G.D.; Hew, B.E.; Waller, D.F.; Holdaway, C.; Jen, M. Amperometric Detection of Bacillus anthracis Spores: A Portable, Low-Cost Approach to the ELISA. Int. J. Electrochem. 2013, 803485. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Q.; Tang, X.; Zhang, W.; Li, P. Development of an Anti-Idiotypic VHH Antibody and Toxin-Free Enzyme Immunoassay for Ochratoxin A in Cereals. Toxins 2019, 11, 280. [Google Scholar] [CrossRef]
- International Council for Harmonisation (ICH) Harmonised Guideline Bioanalytical Method Validation M10 ICH Consensus Guideline Ligand Binding Assay. pp. 42–52. Available online: https://www.fda.gov›media›download (accessed on 1 November 2019).
- EC. Commission Regulation (EC) No 1881/2006. Off. J. Eur. Union 2006, L364, 5–24. [Google Scholar]



{kind=link}
{kind=link}
| Variable | Pristine Protocol Deduced from [19] | Conditions Considered in This Work 1 |
|---|---|---|
| volume of standard/sample | 100 µL | 25, 50, 100 µL |
| Dilution factor of methanol extract | 1 + 1 | 1 + 0, 1 + 1, 1 + 3 |
| Buffer for diluting AFB1-HRP | PBST pH 7.4 | PBS/T pH 5.0, 6.0, 7.4 MES/T pH 6.0 phosphate/citrate/T pH 6.0 Tris/T pH 7.4, 8.5 |
| Washing solution composition | 0.3 M NaCl + Tween 20 | 0.05% Tween 20, 0.3 M NaCl/T, PBS/T pH 7.4, PBS/T pH 5.0 |
| Time of reaction | 15′ + 15′ | 15′ + 15′, 25′ + 15′, 15′ + 25′ |
| Parameter | Mean ± SD |
|---|---|
| Bmax (UA) | 1.7 ± 0.2 |
| Bmin (UA) | 0.08 ± 0.01 |
| IC50 (ng mL−1) | 0.8 ± 0.1 |
| Slope | −1.26 ± 0.05 |
| Method | Definition of LOD | LOD (ng mL−1) | Definition of ROQ | ROQ (ng mL−1) | Ref. |
|---|---|---|---|---|---|
| Signal-to-noise ratio | B0–3sd0 | 0.2 | linearity (y vs log x) | 0.2–2.5 | [22,23] |
| Bmax inhibition | IC10 | 0.12 | IC20–IC80 | 0.15–4 | [24,25,26,27] |
| Error profile | RSD% = 50% | 0.2 | RSD% = 50% | 0.2–14 | [28] |
| Back-calculation | Inaccuracy = 25% | 0.35 | Inaccuracy = 20% | 0.4–2 | [29] |
| Sample Id # | Enzyme Immunoassay | HPLC-MS/MS | ||
|---|---|---|---|---|
| AFB1 ± SD (ng g−1) | ME a (%) | AFB1 ± SD (ng g−1) | ME a (%) | |
| NH-1 | 12.1 ± 0.9 | 86 | <LOD c | 74 |
| NH-2 | 14.8 ± 2.6 | 111 | <LOD c | 91 |
| GA-1 | <LOD b | 118 | <LOD c | 81 |
| GA-2 | 8.7 ± 0.1 | 117 | <LOD c | 96 |
| WA-1 | <LOD b | 104 | <LOD c | 103 |
| WA-2 | <LOD b | 116 | <LOD c | 123 |
| EJ-1 | <LOD b | 102 | <LOD c | 123 |
| EJ-2 | <LOD b | 103 | <LOD c | 118 |
| VW | 13.8 ± 0.2 | 118 | <LOD c | 99 |
| AF | <LOD | 78 | <LOD c | 119 |
| GS | <LOD | 136 | <LOD c | 91 |
| DP | 13.4 ± 1.8 | 86 | <LOD c | 92 |
| BE | 11.5 ± 1.1 | 126 | <LOD c | 83 |
| LE | <LOD | 98 | <LOD c | 85 |
| AH | <LOD | 116 | <LOD c | 100 |
| JA | 17.7 ± 0.2 | - d | - d | - d |
| DI | 9.7 ± 0.9 | - d | - d | - d |
| SO | 8.6 ± 0.4 | - d | - d | - d |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Nardo, F.; Cavalera, S.; Baggiani, C.; Chiarello, M.; Pazzi, M.; Anfossi, L. Enzyme Immunoassay for Measuring Aflatoxin B1 in Legal Cannabis. Toxins 2020, 12, 265. https://doi.org/10.3390/toxins12040265
Di Nardo F, Cavalera S, Baggiani C, Chiarello M, Pazzi M, Anfossi L. Enzyme Immunoassay for Measuring Aflatoxin B1 in Legal Cannabis. Toxins. 2020; 12(4):265. https://doi.org/10.3390/toxins12040265
Chicago/Turabian StyleDi Nardo, Fabio, Simone Cavalera, Claudio Baggiani, Matteo Chiarello, Marco Pazzi, and Laura Anfossi. 2020. "Enzyme Immunoassay for Measuring Aflatoxin B1 in Legal Cannabis" Toxins 12, no. 4: 265. https://doi.org/10.3390/toxins12040265
APA StyleDi Nardo, F., Cavalera, S., Baggiani, C., Chiarello, M., Pazzi, M., & Anfossi, L. (2020). Enzyme Immunoassay for Measuring Aflatoxin B1 in Legal Cannabis. Toxins, 12(4), 265. https://doi.org/10.3390/toxins12040265