Aspartic Acid Isomerization Characterized by High Definition Mass Spectrometry Significantly Alters the Bioactivity of a Novel Toxin from Poecilotheria

Abstract

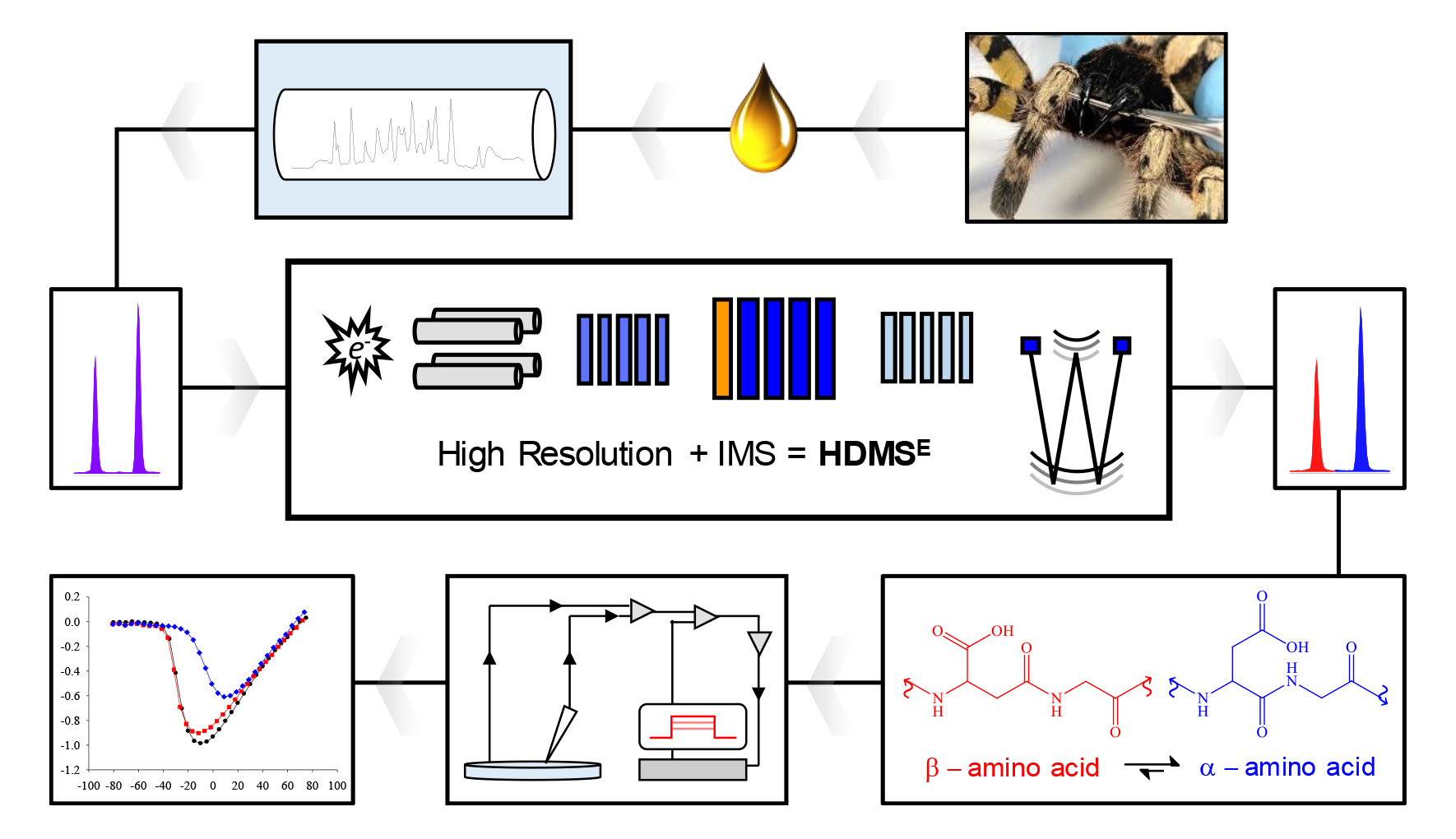
1. Introduction

2. Results
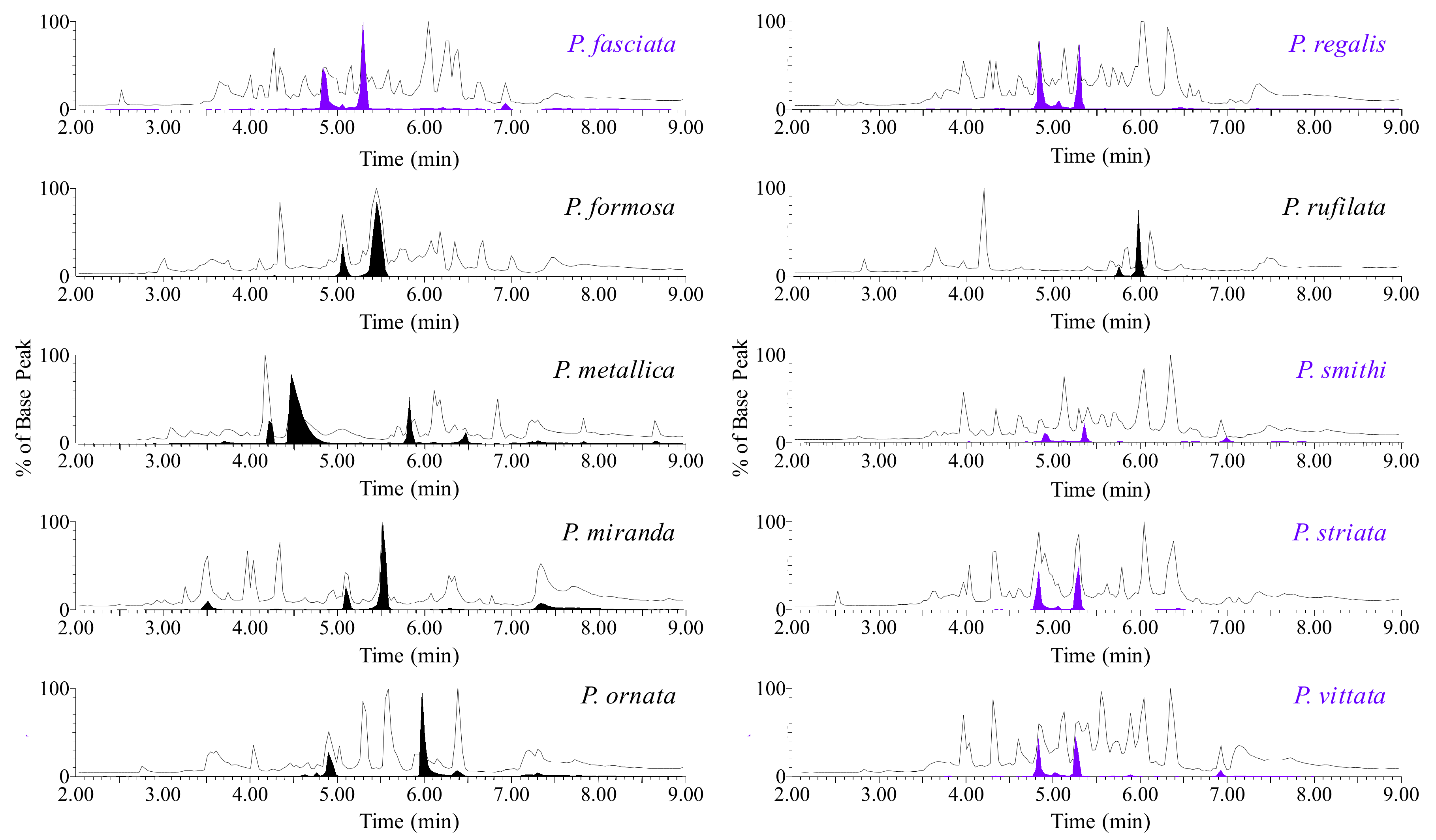
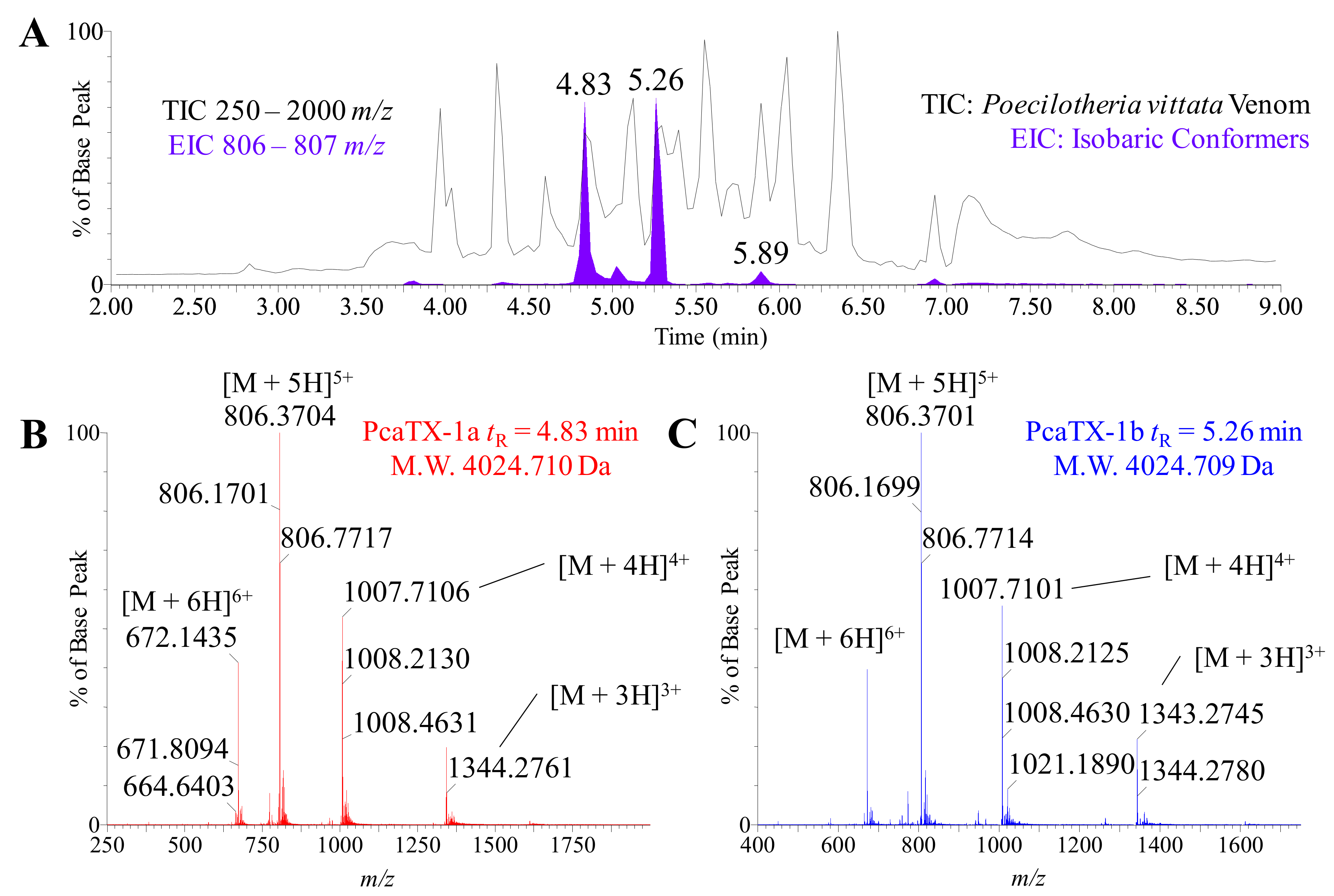
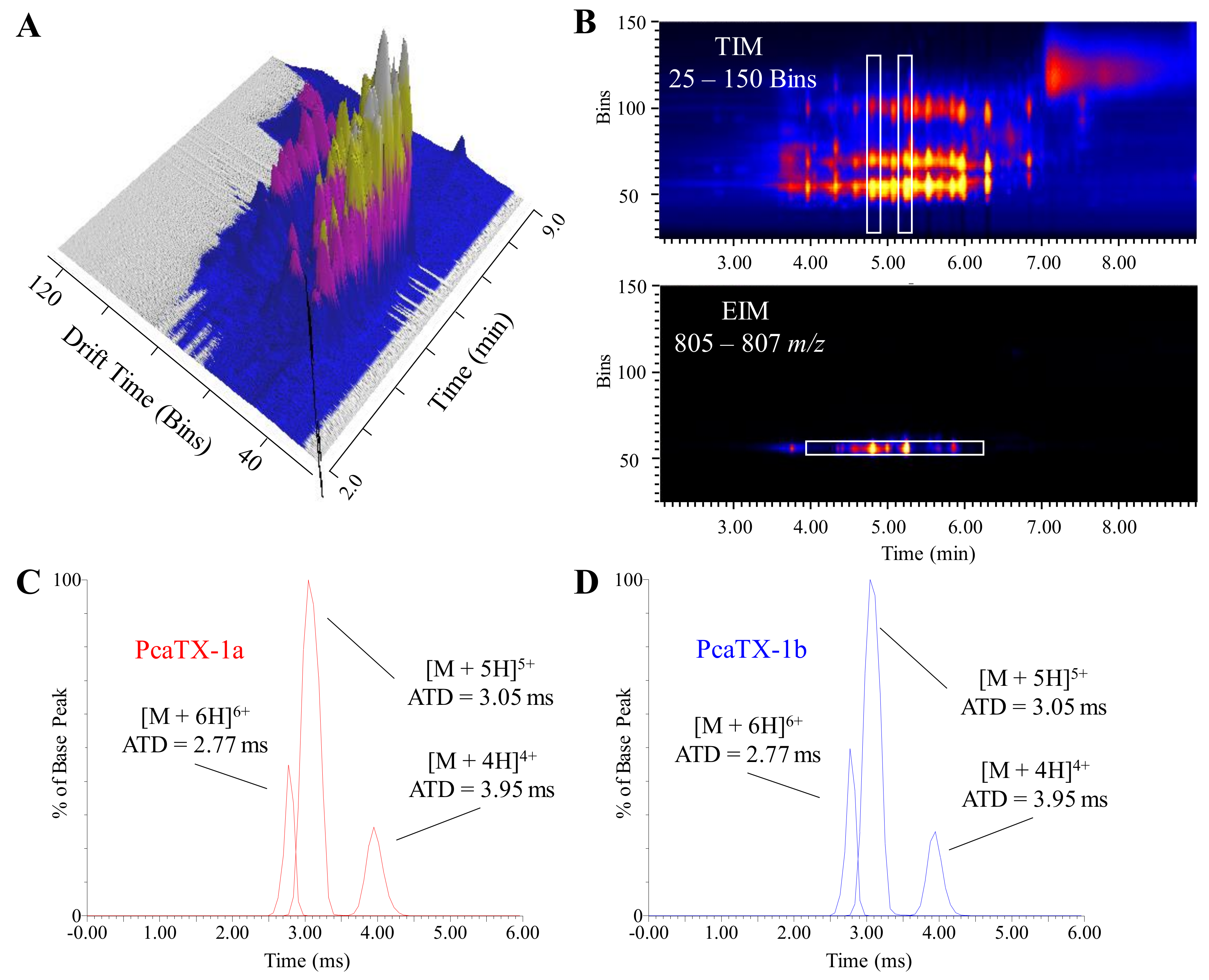
2.1. Toxin Selection
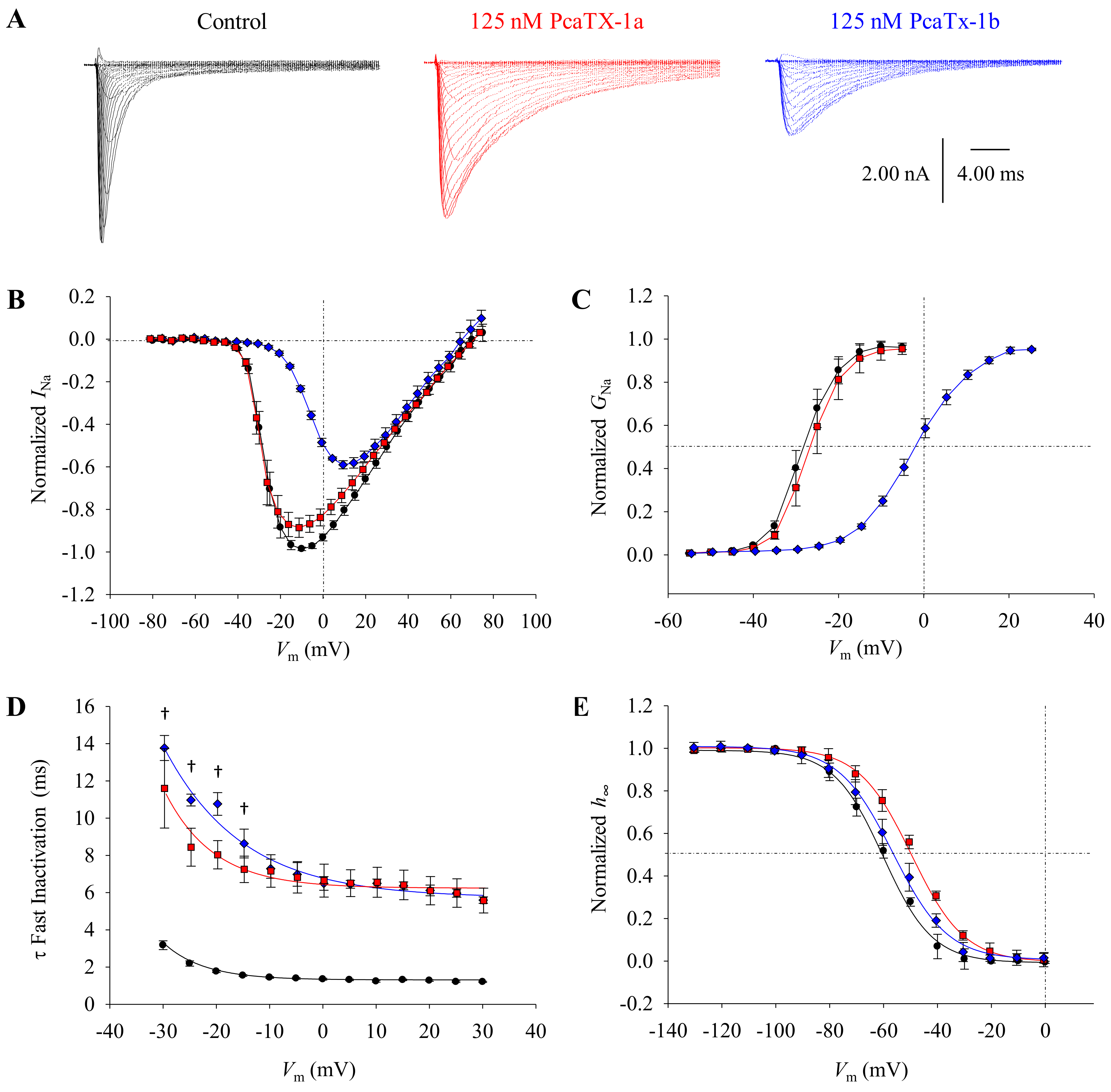
2.2. Effect of PcaTX-1 Conformers on Nav1.7 Currents
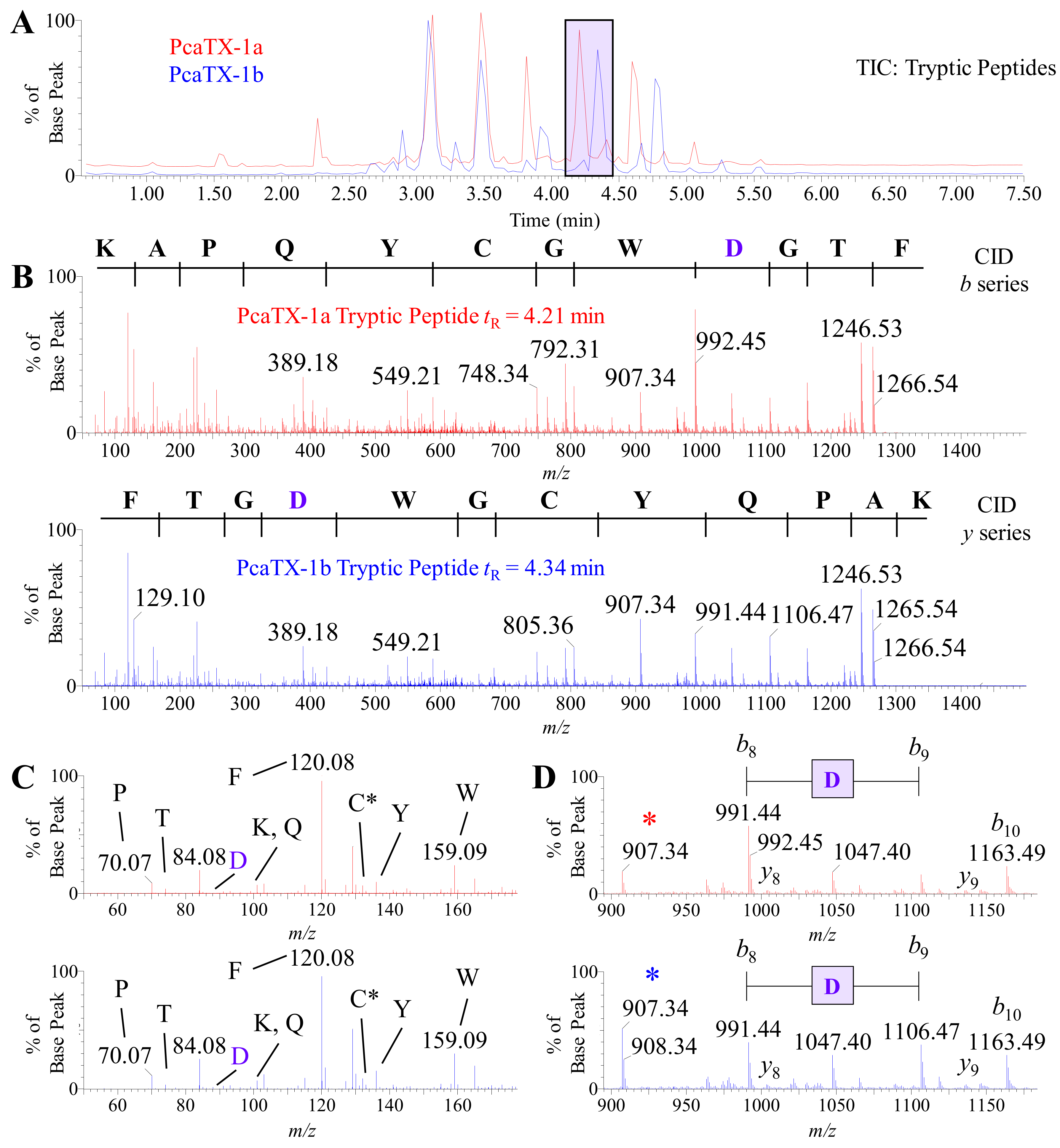
2.3. Preliminary de novo Sequencing of PcaTX-1a and PcaTX-1b by CID
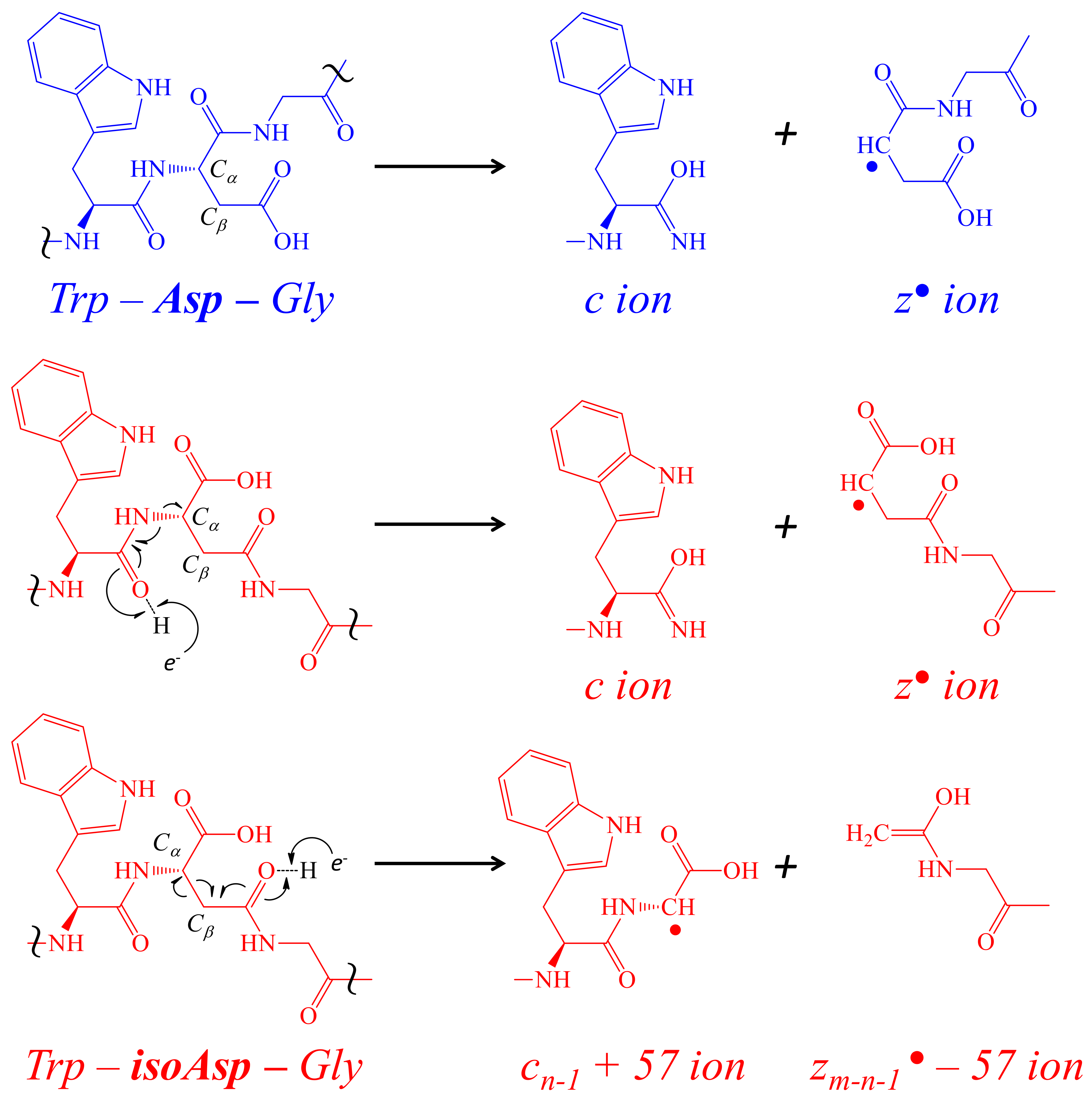
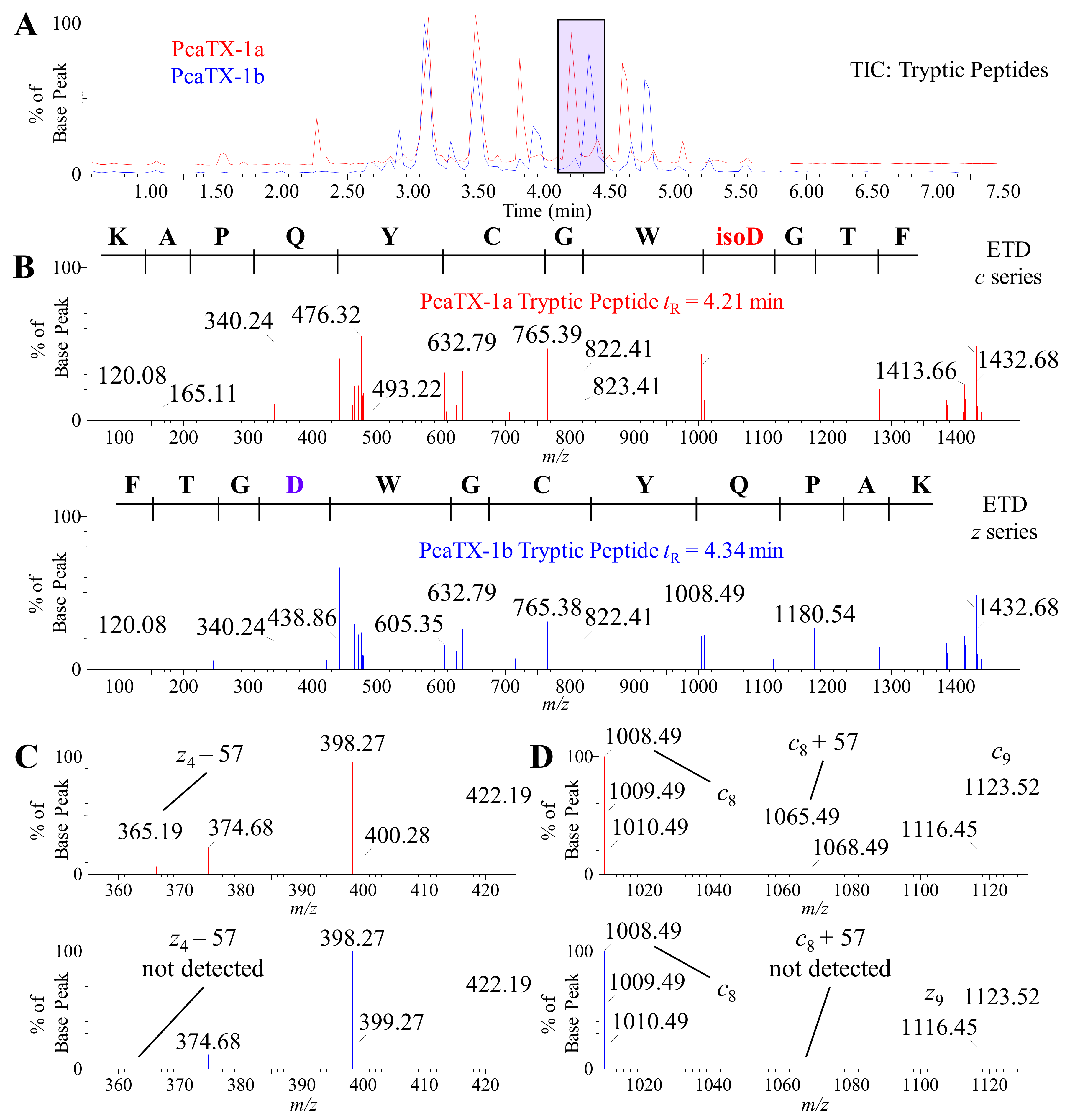
2.4. Confirmatory de novo Sequencing of PcaTX-1a and PcaTX-1b by ETD
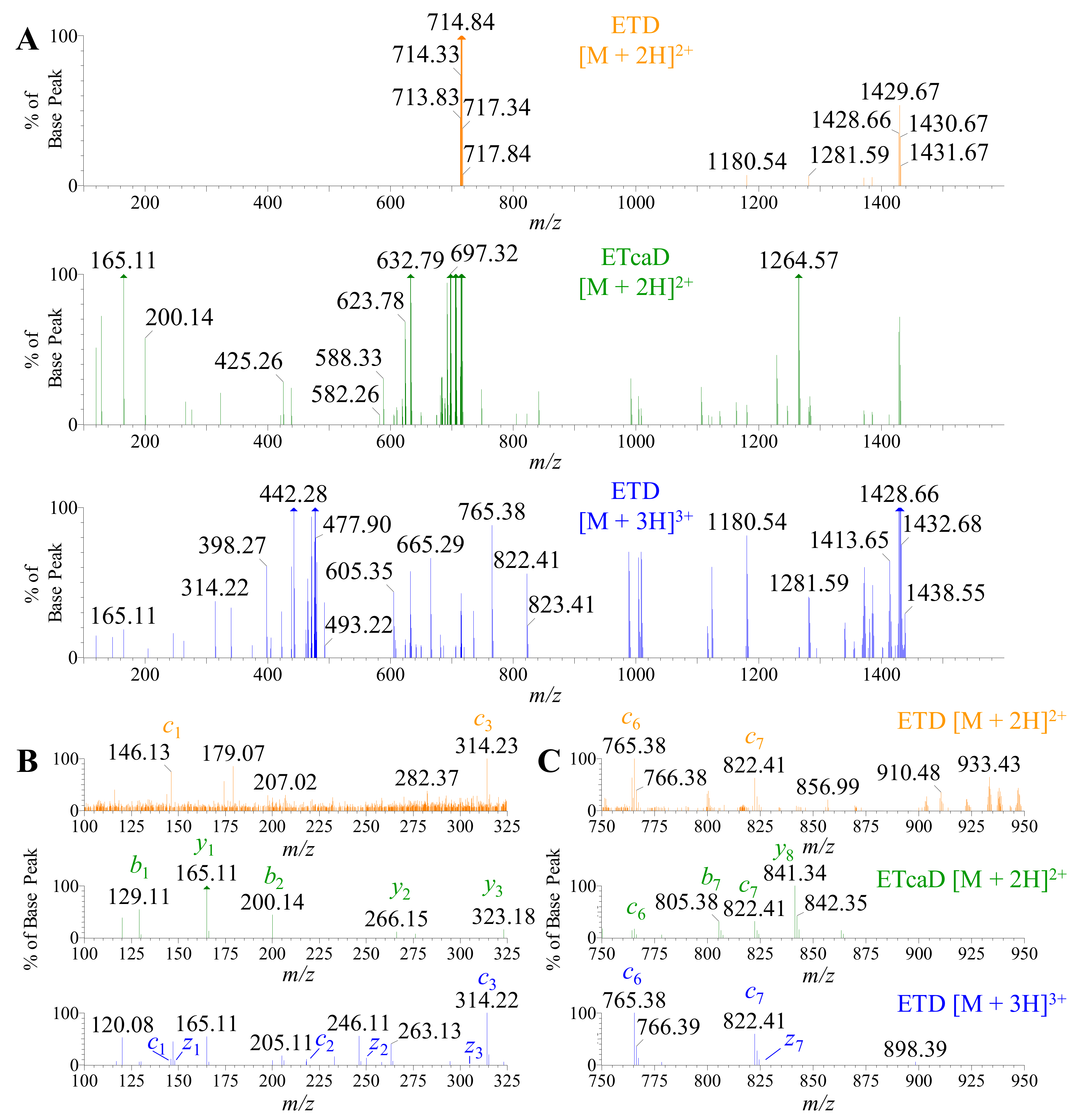
2.5. Using ETD vs. ETcaD
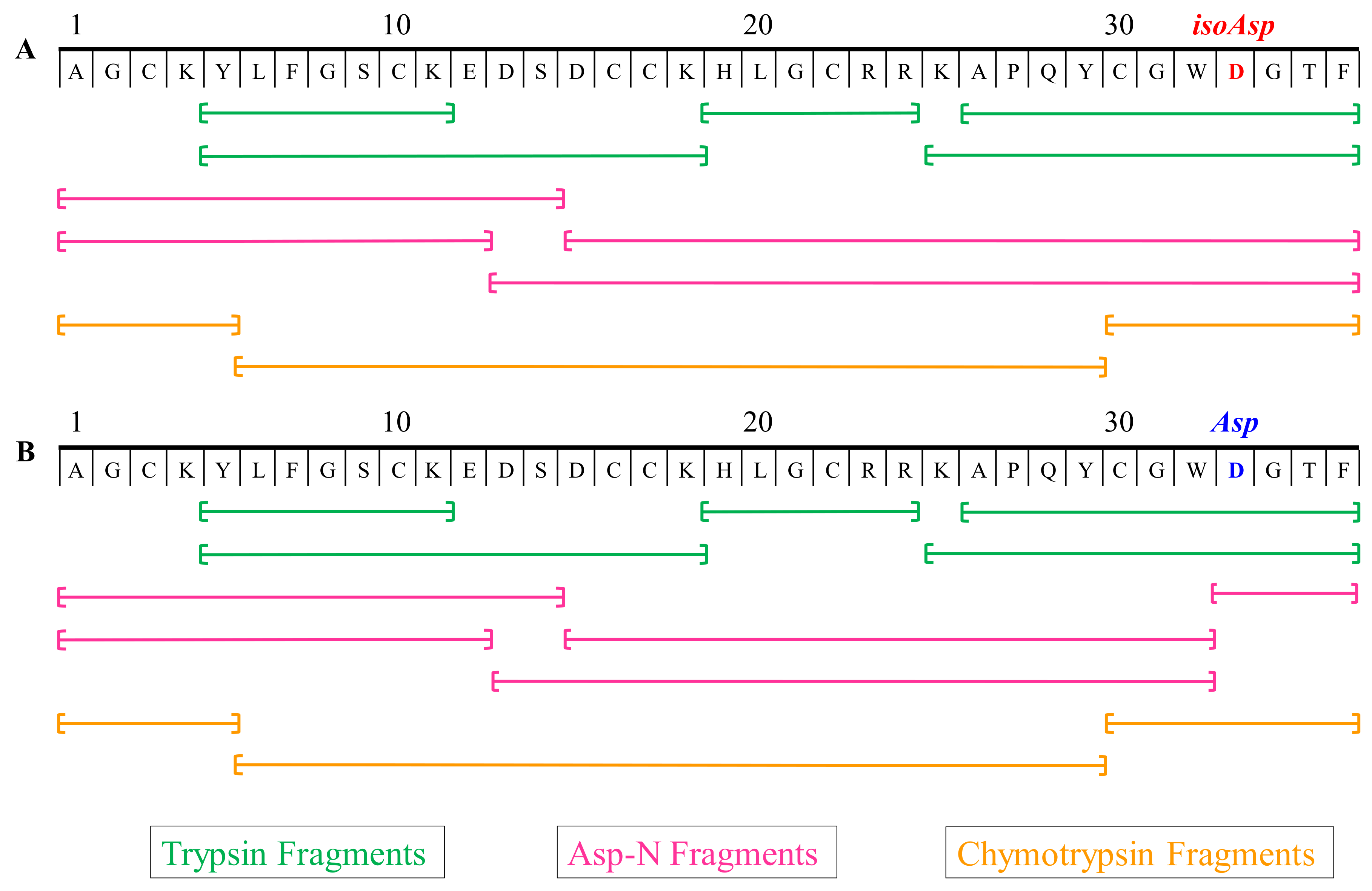
2.6. Multiple Enzymes Digestion Products
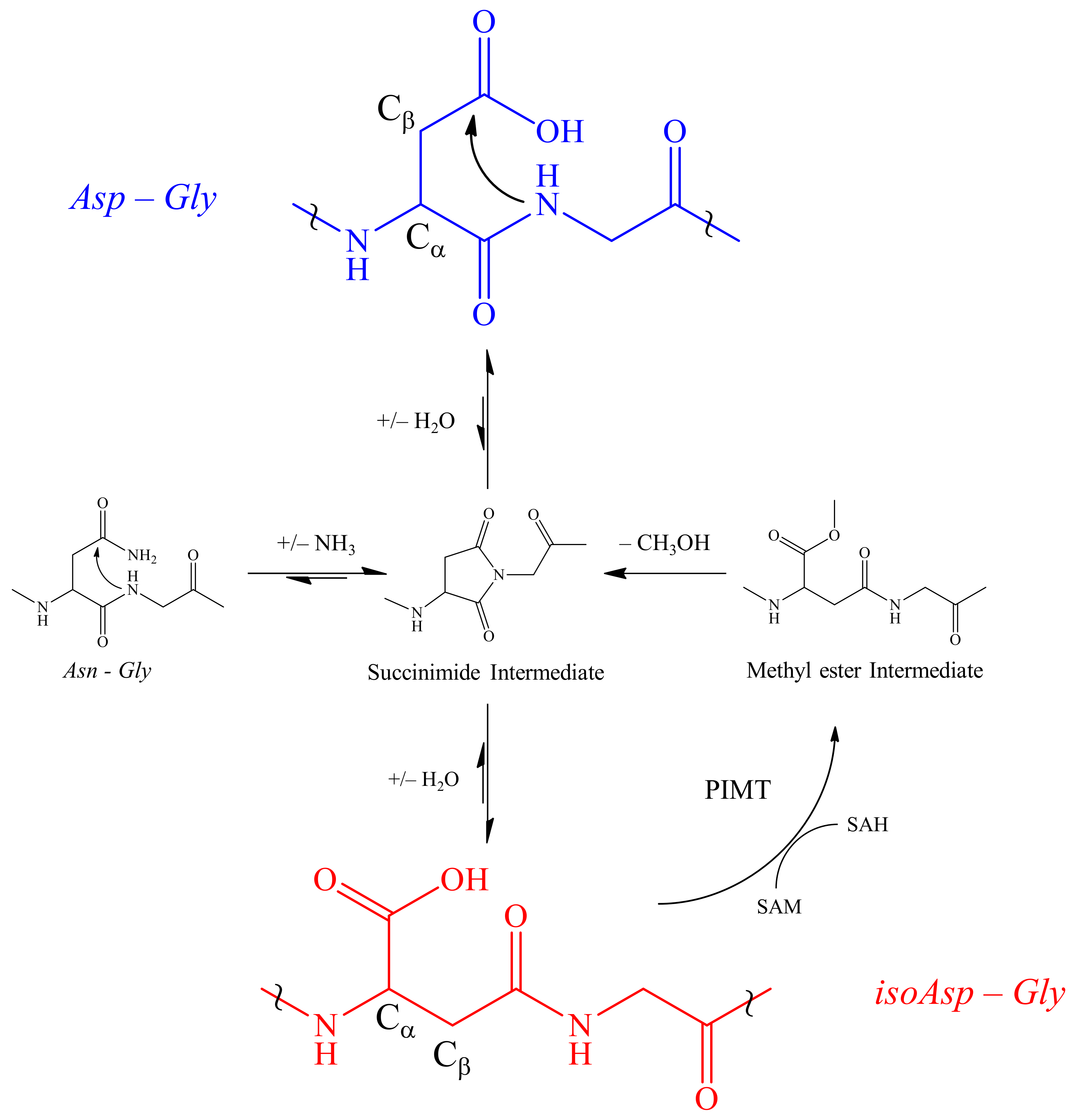
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Reagents and Chemicals
5.2. Venom Collection and Preparation
5.3. Size Exclusion Chromatography (SEC) Purifications
5.4. Reversed-Phase Chromatography (RPC) Purifications
5.5. Reversed-Phase Chromatography (RPC) Determinations
5.6. Toxin Digestions and Modifications
5.7. Accurate Mass Determinations
5.8. Collision Induced Dissociation (CID) and Electron Transfer Dissociation (ETD)
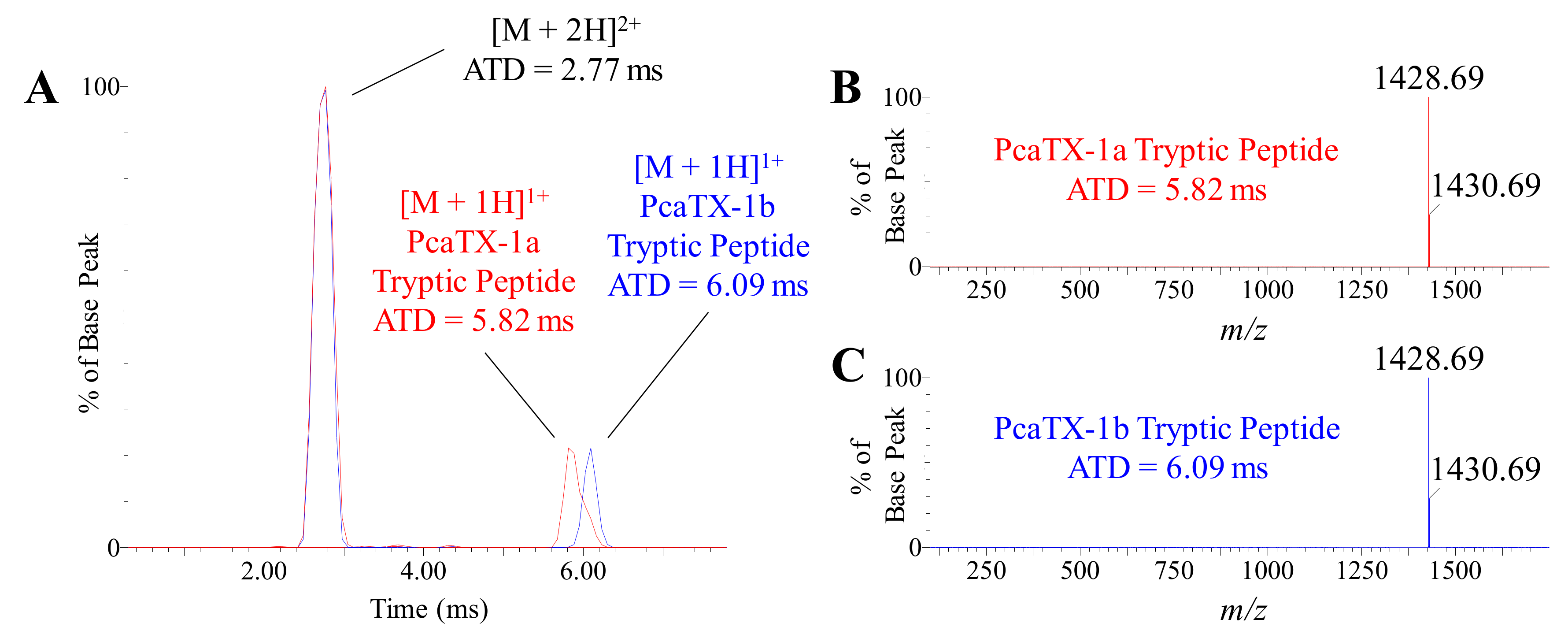
5.9. Traveling Wave Ion Mobility Spectrometry (TWIMS)
5.10. Cell Culture
5.11. Whole-Cell Recording
5.12. Data Analysis and Statistics
5.13. Accession Number
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beneski, D.A.; Catterall, W.A. Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc. Natl. Acad. Sci. USA 1980, 77, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure. function and pathophysiology J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [PubMed]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4, 207. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. Ion Channels and Disease: Channelopathies; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Camerino, D.C.; Desaphy, J.F.; Tricarico, D.; Pierno, S.; Liantonio, A. Therapeutic approaches to ion channel diseases. Adv. Genet. 2008, 64, 81–145. [Google Scholar] [PubMed]
- Kew, J.N.C.; Davies, C.H. Ion. channels: From Structure to Function, 2nd ed.; Oxford University Press: Oxford, UK; New York, NY, USA, 2010. [Google Scholar]
- Moss, S.J.; Henley, J. Receptor and Ion-Channel Trafficking: Cell Biology of Ligand-Gated and Voltage-Sensitive Ion Channels (Molecular and Cellular Neurobiology); Oxford University Press: Oxford, UK; New York, NY, USA, 2002. [Google Scholar]
- Thiel, M.; Zourelidis, C.; Chambers, J.D.; von Andrian, U.H.; Arfors, K.E.; Messmer, K.; Peter, K. Expression of beta 2-integrins and L-selectin on polymorphonuclear leukocytes in septic patients. Eur. Surg. Res. 1997, 29, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Chapman, M.L.; Marron, B.E.; Prime, R.; Storer, R.I.; Swain, N.A. Recent progress in sodium channel modulators for pain. Bioorg. Med. Chem. Lett. 2014, 24, 3690–3699. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.C.; Lewis, R.J. Sodium channels and pain: From toxins to therapies. Br. J. Pharmacol. 2018, 175, 2138–2157. [Google Scholar] [CrossRef]
- Lai, J.; Hunter, J.C.; Porreca, F. The role of voltage-gated sodium channels in neuropathic pain. Curr. Opin. Neurobiol. 2003, 13, 291–297. [Google Scholar] [CrossRef]
- Luiz, A.P.; Wood, J.N. Sodium Channels in Pain and Cancer: New Therapeutic Opportunities. Adv. Pharmacol. 2016, 75, 153–178. [Google Scholar]
- Savio-Galimberti, E.; Gollob, M.H.; Darbar, D. Voltage-gated sodium channels: Biophysics, pharmacology, and related channelopathies. Front. Pharmacol. 2012, 3, 124. [Google Scholar] [CrossRef]
- Faber, C.G.; Lauria, G.; Merkies, I.S.; Cheng, X.; Han, C.; Ahn, H.S.; Persson, A.K.; Hoeijmakers, J.G.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Kurban, M.; Wajid, M.; Shimomura, Y.; Christiano, A.M. A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology 2010, 221, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Kyle, D.J.; Ilyin, V.I. Sodium channel blockers. J. Med. Chem. 2007, 50, 2583–2588. [Google Scholar] [CrossRef]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef]
- Zhang, A.H.; Sharma, G.; Undheim, E.A.B.; Jia, X.; Mobli, M. A complicated complex: Ion channels, voltage sensing, cell membranes and peptide inhibitors. Neurosci. Lett. 2018, 679, 35–47. [Google Scholar] [CrossRef]
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology. 2017, 127, 87–108. [Google Scholar] [CrossRef]
- Israel, M.R.; Tay, B.; Deuis, J.R.; Vetter, I. Sodium Channels and Venom Peptide Pharmacology. Adv. Pharmacol. 2017, 79, 67–116. [Google Scholar]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Ménez, A. Perspectives in Molecular Toxinology; John Wiley: New York, NY, USA, 2002. [Google Scholar]
- Wu, Y.; Ma, H.; Zhang, F.; Zhang, C.; Zou, X.; Cao, Z. Selective Voltage-Gated Sodium Channel Peptide Toxins from Animal Venom: Pharmacological Probes and Analgesic Drug Development. ACS Chem. Neurosci. 2018, 9, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar] [CrossRef]
- Agwa, A.J.; Lawrence, N.; Deplazes, E.; Cheneval, O.; Chen, R.M.; Craik, D.J.; Schroeder, C.I.; Henriques, S.T. Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. Biochim. Biophys. Acta Biomembr. 2017, 1859, 835–844. [Google Scholar] [CrossRef]
- Moyer, B.D.; Murray, J.K.; Ligutti, J.; Andrews, K.; Favreau, P.; Jordan, J.B.; Lee, J.H.; Liu, D.; Long, J.; Sham, K.; et al. Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS ONE 2018, 13, e0196791. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Dyubankova, N.; Lescrinier, E.; Herdewijn, P.; Tytgat, J. Design of bioactive peptides from naturally occurring mu-conotoxin structures. J. Biol. Chem. 2012, 287, 31382–31392. [Google Scholar] [CrossRef]
- Compton, P.D.; Kelleher, N.L.; Gunawardena, J. Estimating the Distribution of Protein Post-Translational Modification States by Mass Spectrometry. J. Proteome Res. 2018, 17, 2727–2734. [Google Scholar] [CrossRef]
- Bossu, J.L.; Feltz, A.; Rodeau, J.L.; Tanzi, F. Voltage-dependent transient calcium currents in freshly dissociated capillary endothelial cells. FEBS Lett. 1989, 255, 377–380. [Google Scholar] [CrossRef]
- Escoubas, P.; Rash, L. Tarantulas: Eight-legged pharmacists and combinatorial chemists. Toxicon 2004, 43, 555–574. [Google Scholar] [CrossRef]
- Hungerford, J.M.; Committee on Natural Toxins and Food Allergens. Marine and freshwater toxins. J. AOAC Int. 2006, 89, 248–269. [Google Scholar] [CrossRef] [PubMed]
- Wright, Z.V.F.; McCarthy, S.; Dickman, R.; Reyes, F.E.; Sanchez-Martinez, S.; Cryar, A.; Kilford, I.; Hall, A.; Takle, A.K.; Topf, M.; et al. The Role of Disulfide Bond Replacements in Analogues of the Tarantula Toxin ProTx-II and Their Effects on Inhibition of the Voltage-Gated Sodium Ion Channel Nav1.7. J. Am. Chem. Soc. 2017, 139, 13063–13075. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, C.; Boudier, C.; Bermudez, I.; Bieth, J.G.; Luu, B.; Hietter, H. Serine protease inhibition by insect peptides containing a cysteine knot and a triple-stranded beta-sheet. J. Biol Chem. 1995, 270, 25514–25519. [Google Scholar] [CrossRef] [PubMed]
- Kastin, A.J. Handbook of Biologically active Peptides, 2nd ed.; Elsevier/AP: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Kumar, D.; Mains, R.E.; Eipper, B.A. 60 YEARS OF POMC: From POMC and alpha-MSH to PAM, molecular oxygen, copper, and vitamin C. J. Mol. Endocrinol. 2016, 56, T63–T76. [Google Scholar] [CrossRef]
- Kumar, D.; Strenkert, D.; Patel-King, R.S.; Leonard, M.T.; Merchant, S.S.; Mains, R.E.; King, S.M.; Eipper, B.A. A bioactive peptide amidating enzyme is required for ciliogenesis. Elife 2017, 6, e25728. [Google Scholar] [CrossRef]
- Benkhadir, K.; Kharrat, R.; Cestele, S.; Mosbah, A.; Rochat, H.; El Ayeb, M.; Karoui, H. Molecular cloning and functional expression of the alpha-scorpion toxin BotIII: Pivotal role of the C-terminal region for its interaction with voltage-dependent sodium channels. Peptides 2004, 25, 151–161. [Google Scholar] [CrossRef]
- Billen, B.; Vassilevski, A.; Nikolsky, A.; Debaveye, S.; Tytgat, J.; Grishin, E. Unique bell-shaped voltage-dependent modulation of Na+ channel gating by novel insect-selective toxins from the spider Agelena orientalis. J. Biol. Chem. 2010, 285, 18545–18554. [Google Scholar] [CrossRef]
- Buczek, O.; Yoshikami, D.; Bulaj, G.; Jimenez, E.C.; Olivera, B.M. Post-translational amino acid isomerization: A functionally important D-amino acid in an excitatory peptide. J. Biol. Chem. 2005, 280, 4247–4253. [Google Scholar] [CrossRef]
- Capasso, S.; Di Cerbo, P. Kinetic and thermodynamic control of the relative yield of the deamidation of asparagine and isomerization of aspartic acid residues. J. Pept. Res. 2000, 56, 382–387. [Google Scholar] [CrossRef]
- Clarke, S. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int. J. Pept. Protein Res. 1987, 30, 808–821. [Google Scholar] [CrossRef]
- Andreev-Andrievskiy, A.; Popova, A.; Lagereva, E.; Osipov, D.; Berkut, A.; Grishin, E.; Vassilevski, A. Pharmacological analysis of Poecilotheria spider venoms in mice provides clues for human treatment. Toxicon 2017, 138, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Corzo, G.; Whiteley, B.J.; Celerier, M.L.; Nakajima, T. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and high-performance liquid chromatography study of quantitative and qualitative variation in tarantula spider venoms. Rapid Commun. Mass Spectrom. 2002, 16, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arredondo, A.; Rodriguez-Rios, L.; Diaz-Pena, L.F.; Vega-Angeles, R. Pharmacological characterization of venoms from three theraphosid spiders: Poecilotheria regalis. Ceratogyrus darlingi and Brachypelma epicureanum. J. Venom. Anim. Toxins Incl. Trop. Dis. 2015, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Oldrati, V.; Koua, D.; Allard, P.M.; Hulo, N.; Arrell, M.; Nentwig, W.; Lisacek, F.; Wolfender, J.L.; Kuhn-Nentwig, L.; Stocklin, R. Peptidomic and transcriptomic profiling of four distinct spider venoms. PLoS ONE 2017, 12, e0172966. [Google Scholar] [CrossRef]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef]
- Shvartsburg, A.A.; Smith, R.D. Fundamentals of traveling wave ion mobility spectrometry. Anal. Chem. 2008, 80, 9689–9699. [Google Scholar] [CrossRef]
- Wang, Y.; Park, K.D.; Salome, C.; Wilson, S.M.; Stables, J.P.; Liu, R.; Khanna, R.; Kohn, H. Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS Chem. Neurosci. 2011, 2, 90–106. [Google Scholar] [CrossRef]
- Bosmans, F.; Rash, L.; Zhu, S.; Diochot, S.; Lazdunski, M.; Escoubas, P.; Tytgat, J. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 2006, 69, 419–429. [Google Scholar] [CrossRef]
- Karoly, R.; Lenkey, N.; Juhasz, A.O.; Vizi, E.S.; Mike, A. Fast- or slow-inactivated state preference of Na+ channel inhibitors: A simulation and experimental study. PLoS Comput. Biol. 2010, 6, e1000818. [Google Scholar] [CrossRef]
- Biemann, K. Appendix Mass values for amino acid residues in peptides. Methods Enzymol. 1990, 193, 888. [Google Scholar]
- Biemann, K. Appendix Nomenclature for peptide fragment ions (positive ions). Methods Enzymol. 1990, 193, 886–887. [Google Scholar]
- Biemann, K. Peptides and proteins: Overview and strategy. Methods Enzymol. 1990, 193, 351–360. [Google Scholar] [PubMed]
- Hohmann, L.J.; Eng, J.K.; Gemmill, A.; Klimek, J.; Vitek, O.; Reid, G.E.; Martin, D.B. Quantification of the compositional information provided by immonium ions on a quadrupole-time-of-flight mass spectrometer. Anal. Chem. 2008, 80, 5596–5606. [Google Scholar] [CrossRef] [PubMed]
- Coon, J.J.; Syka, J.E.; Shabanowitz, J.; Hunt, D.F. Tandem mass spectrometry for peptide and protein sequence analysis. Biotechniques 2005, 38, 519–523. [Google Scholar] [CrossRef]
- Syka, J.E.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef]
- Sargaeva, N.P.; Lin, C.; O’Connor, P.B. Differentiating N-terminal aspartic and isoaspartic acid residues in peptides. Anal. Chem. 2011, 83, 6675–6682. [Google Scholar] [CrossRef]
- Swaney, D.L.; McAlister, G.C.; Wirtala, M.; Schwartz, J.C.; Syka, J.E.; Coon, J.J. Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 2007, 79, 477–485. [Google Scholar] [CrossRef]
- Heissel, S.; Frederiksen, S.J.; Bunkenborg, J.; Hojrup, P. Enhanced trypsin on a budget: Stabilization. purification and high-temperature application of inexpensive commercial trypsin for proteomics applications. PLoS ONE 2019, 14, e0218374. [Google Scholar] [CrossRef]
- Hustoft, H.K.; Reubsaet, L.; Greibrokk, T.; Lundanes, E.; Malerod, H. Critical assessment of accelerating trypsination methods. J. Pharm. Biomed. Anal. 2011, 56, 1069–1078. [Google Scholar] [CrossRef]
- Chan, W.Y.; Chan, T.W.; O’Connor, P.B. Electron transfer dissociation with supplemental activation to differentiate aspartic and isoaspartic residues in doubly charged peptide cations. J. Am. Soc. Mass Spectrom. 2010, 21, 1012–1015. [Google Scholar] [CrossRef]
- Choudhary, G.; Wu, S.L.; Shieh, P.; Hancock, W.S. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J. Proteome Res. 2003, 2, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Dai, S.; Karger, B.L.; Zhou, Z.S. Analysis of isoaspartic Acid by selective proteolysis with Asp-N and electron transfer dissociation mass spectrometry. Anal. Chem. 2010, 82, 7485–7491. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Watson, J.T. A novel methodology for assignment of disulfide bond pairings in proteins. Protein Sci. 1997, 6, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.W.; Jung, S.; Grotzinger, J.; Gelhaus, C.; Leippe, M.; Tholey, A. Determination of disulfide linkages in antimicrobial peptides of the macin family by combination of top-down and bottom-up proteomics. J. Proteom. 2014, 103, 216–226. [Google Scholar] [CrossRef]
- Wu, S.L.; Jiang, H.; Lu, Q.; Dai, S.; Hancock, W.S.; Karger, B.L. Mass spectrometric determination of disulfide linkages in recombinant therapeutic proteins using online LC-MS with electron-transfer dissociation. Anal. Chem. 2009, 81, 112–122. [Google Scholar] [CrossRef]
- Pimenta, A.M.; De Lima, M.E. Small peptides, big world: Biotechnological potential in neglected bioactive peptides from arthropod venoms. J. Pept. Sci. 2005, 11, 670–676. [Google Scholar] [CrossRef]
- von Reumont, B.M.; Campbell, L.I.; Jenner, R.A. Quo vadis venomics? A roadmap to neglected venomous invertebrates. Toxins 2014, 6, 3488–3551. [Google Scholar] [CrossRef]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef]
- Shcherbatko, A.; Rossi, A.; Foletti, D.; Zhu, G.; Bogin, O.; Galindo Casas, M.; Rickert, M.; Hasa-Moreno, A.; Bartsevich, V.; Crameri, A.; et al. Engineering Highly Potent and Selective Microproteins against Nav1.7 Sodium Channel for Treatment of Pain. J. Biol. Chem. 2016, 291, 13974–13986. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural Products as a Foundation for Drug Discovery. Curr. Protoc. Pharmacol. 2009, 46, 9–11. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural Products as a Foundation for Drug Discovery. Curr. Protoc. Pharmacol. 2019, 86, e67. [Google Scholar] [CrossRef]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Narahashi, T. Neuroreceptors and ion channels as the basis for drug action: Past, present, and future. J. Pharmacol. Exp. Ther. 2000, 294, 1–26. [Google Scholar] [PubMed]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed]
- DeGraan-Weber, N.; Zhang, J.; Reilly, J.P. Distinguishing Aspartic and Isoaspartic Acids in Peptides by Several Mass Spectrometric Fragmentation Methods. J. Am. Soc. Mass Spectrom. 2016, 27, 2041–2053. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.R.; Fanelus, I. Damaged proteins bearing L-isoaspartyl residues and aging: A dynamic equilibrium between generation of isomerized forms and repair by PIMT. Curr. Aging Sci. 2011, 4, 8–18. [Google Scholar] [CrossRef]
- Fujii, N.; Sakaue, H.; Sasaki, H.; Fujii, N. A rapid, comprehensive liquid chromatography-mass spectrometry (LC-MS)-based survey of the Asp isomers in crystallins from human cataract lenses. J. Biol. Chem. 2012, 287, 39992–40002. [Google Scholar] [CrossRef]
- Liu, S.; Moulton, K.R.; Auclair, J.R.; Zhou, Z.S. Mildly acidic conditions eliminate deamidation artifact during proteolysis: Digestion with endoprotease Glu-C at pH 4.5. Amino Acids 2016, 48, 1059–1067. [Google Scholar] [CrossRef]
- Reissner, K.J.; Aswad, D.W. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 2003, 60, 1281–1295. [Google Scholar] [CrossRef]
- Johnson, B.A.; Murray, E.D., Jr.; Clarke, S.; Glass, D.B.; Aswad, D.W. Protein carboxyl methyltransferase facilitates conversion of atypical L-isoaspartyl peptides to normal L-aspartyl peptides. J. Biol. Chem. 1987, 262, 5622–5629. [Google Scholar]
- Ulbricht, W. Sodium channel inactivation: Molecular determinants and modulation. Physiol. Rev. 2005, 85, 1271–1301. [Google Scholar] [CrossRef]
- Masuda, A.; Dohmae, N. Amino acid analysis of sub-picomolar amounts of proteins by precolumn fluorescence derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Biosci. Trends 2011, 5, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.A.; De Antonis, K.M. Applications of amino acid derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Analysis of feed grains, intravenous solutions and glycoproteins. J. Chromatogr. A 1994, 661, 25–34. [Google Scholar] [CrossRef]
- Pierce Biotechnology. Mass Spectrometry-Grade Endoproteinases; Thermo Fisher Scientific Inc.: Waltham, MA, USA, 2013. [Google Scholar]
- Eckers, C.; Laures, A.M.; Giles, K.; Major, H.; Pringle, S. Evaluating the utility of ion mobility separation in combination with high-pressure liquid chromatography/mass spectrometry to facilitate detection of trace impurities in formulated drug products. Rapid Commun. Mass Spectrom. 2007, 21, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.; Pringle, S.D.; Worthington, K.R.; Little, D.; Wildgoose, J.L.; Bateman, R.H. Applications of a travelling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun. Mass Spectrom. 2004, 18, 2401–2414. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Rikli, H.G.; Schmidt, J.O.; Evans, M.S. A reexamination of poneratoxin from the venom of the bullet ant Paraponera clavata. Peptides 2017, 98, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.F.; Hall, Z.; Giles, K.; Hoyes, J.; Robinson, C.V.; Ruotolo, B.T. Collision cross sections of proteins and their complexes: A calibration framework and database for gas-phase structural biology. Anal. Chem. 2010, 82, 9557–9565. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | PcaTX-1a | PcaTX-1b | pANOVA |
|---|---|---|---|---|
| I V½ (mV) | −32.9 ± 0.889 | −33.6 ± 0.683 | −14.5± 0.592 *,† | <0.000001 |
| I Vrev (mV) | 67.7 ± 0.667 | 70.6 ± 0.384 | 68.6 ± 1.27 | 0.08 |
| G V½ (mV) | −28.6 ± 0.861 | −28.4 ± 0.768 | −6.34 ± 0.490 *,† | <0.0000001 |
| G k | 3.34 ± 0.297 | 3.38 ± 0.482 | 5.47 ± 0.332 *,† | 0.006 |
| h∞V½ (mV) | −58.6 ± 1.18 | −48.6 ± 1.16* | −54.1 ± 0.889 | 0.02 |
| h∞ k | 9.48 ± 0.176 | 9.47 ± 0.355 | 9.55 ± 0.367 | 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, S.R.; Rikli, H.G. Aspartic Acid Isomerization Characterized by High Definition Mass Spectrometry Significantly Alters the Bioactivity of a Novel Toxin from Poecilotheria. Toxins 2020, 12, 207. https://doi.org/10.3390/toxins12040207
Johnson SR, Rikli HG. Aspartic Acid Isomerization Characterized by High Definition Mass Spectrometry Significantly Alters the Bioactivity of a Novel Toxin from Poecilotheria. Toxins. 2020; 12(4):207. https://doi.org/10.3390/toxins12040207
Chicago/Turabian StyleJohnson, Stephen R., and Hillary G. Rikli. 2020. "Aspartic Acid Isomerization Characterized by High Definition Mass Spectrometry Significantly Alters the Bioactivity of a Novel Toxin from Poecilotheria" Toxins 12, no. 4: 207. https://doi.org/10.3390/toxins12040207
APA StyleJohnson, S. R., & Rikli, H. G. (2020). Aspartic Acid Isomerization Characterized by High Definition Mass Spectrometry Significantly Alters the Bioactivity of a Novel Toxin from Poecilotheria. Toxins, 12(4), 207. https://doi.org/10.3390/toxins12040207