Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways

Abstract
1. Introduction
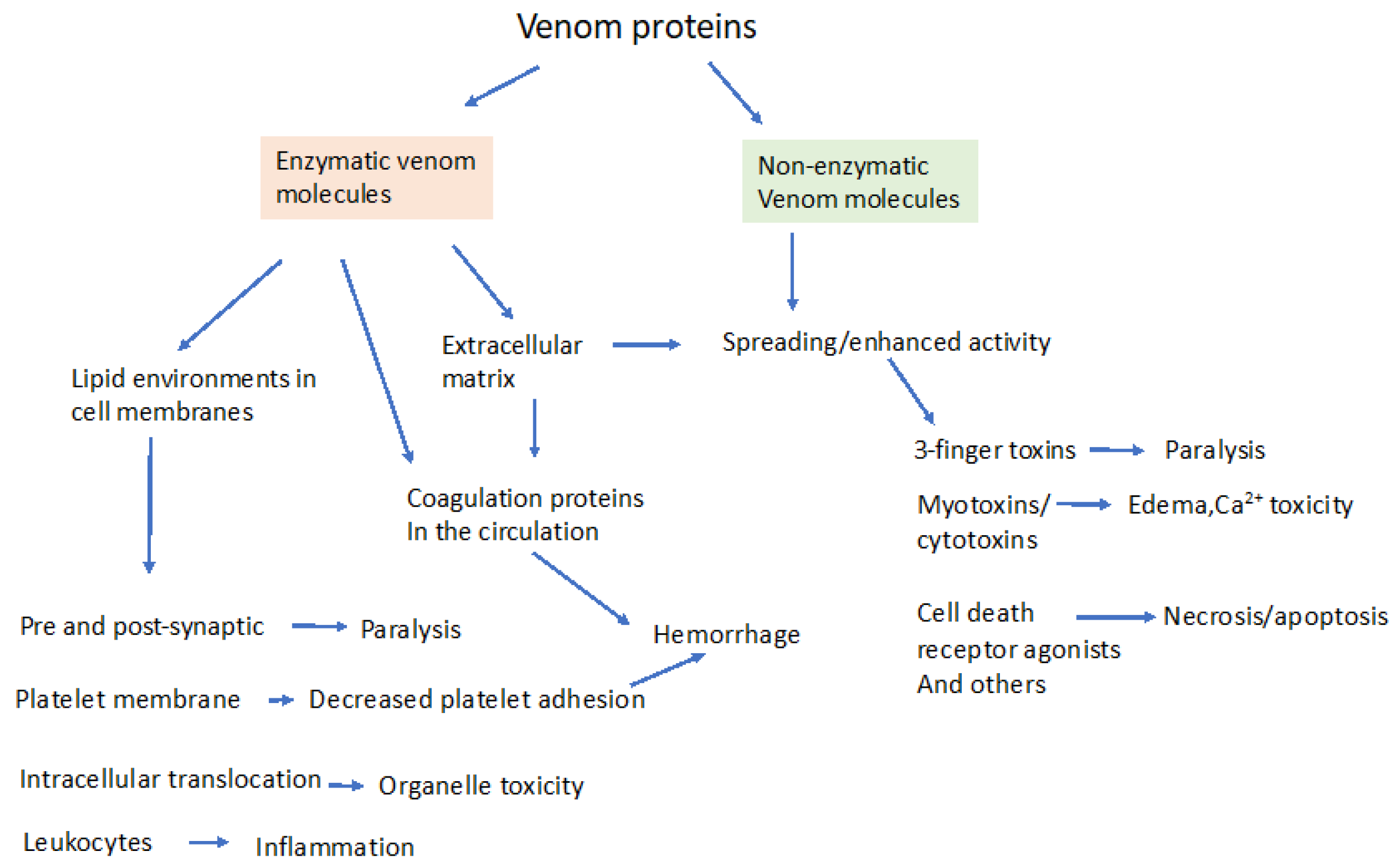
2. Enzymatic and Non-Enzymatic Components of Venom and Where They Act
2.1. Venom Translocation from Circulation to Interstitial Compartment
2.2. Venom Binding or Interaction with Cell Surface Receptors
2.3. Venom Transport into the Intracellular Compartment
3. Molecular Effectors in Elapid and Viper Venoms
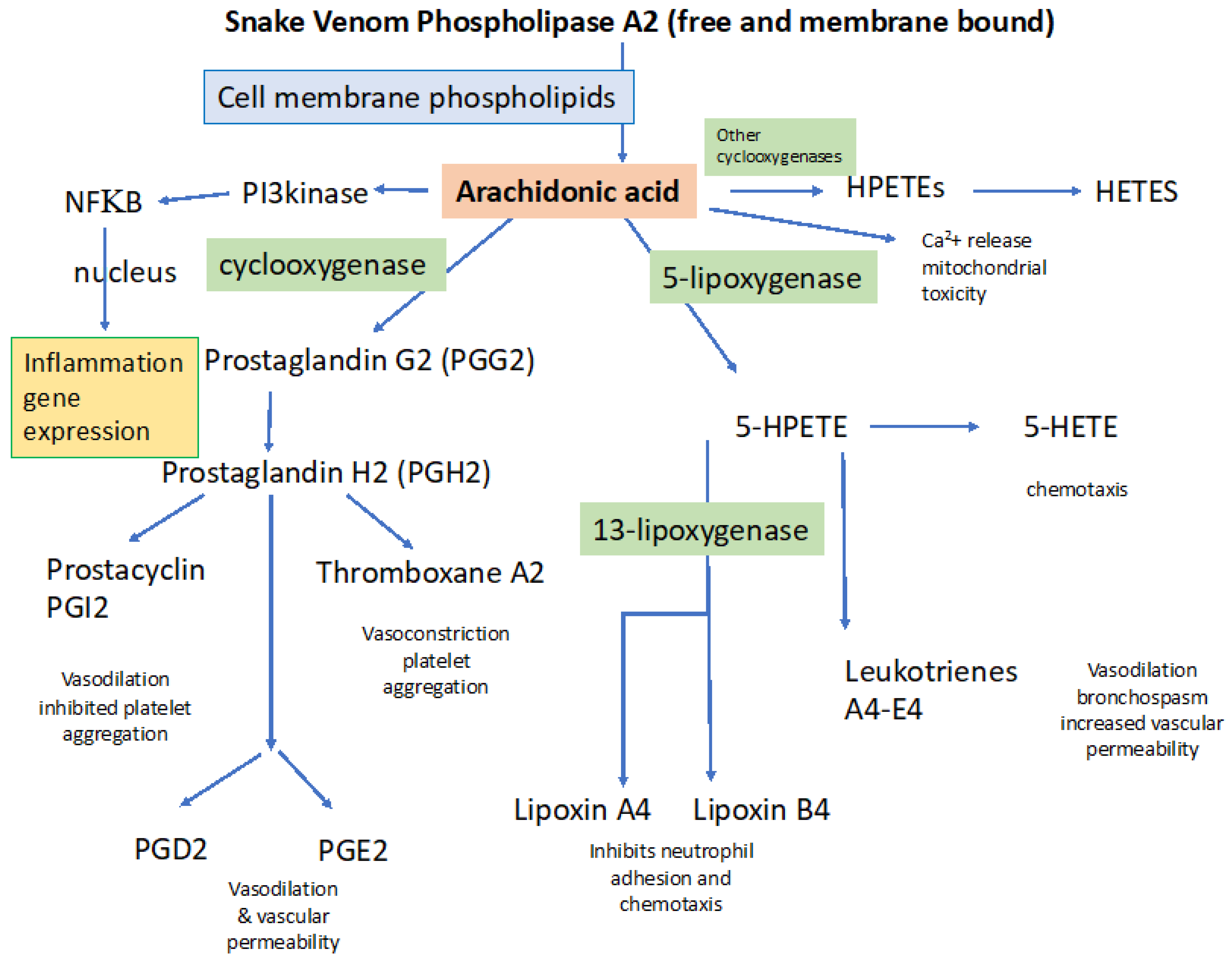
3.1. Snake Venom Phospholipases (svPLA2)
3.2. Snake Venom Metalloprotease (svMP)
3.3. Snake Venon Hyaluronidases
3.4. Other Directly Cytotoxic Proteins
3.5. Non-Enzymatic 3-Finger Toxins (3FTx, -Neurotoxins)
4. Prey Response to Venoms: How Venoms Co-Opt Signaling Pathways to Produce Toxicity
4.1. Inflammation and Inflammation-Mediated Cytotoxicity
4.1.1. General Processes Involved in Activating Inflammation: Importance of PLA2
4.1.2. Inflammation Underlies Increased Vascular Permeability and Tissue Edema
4.2. Coagulation Disorders
4.2.1. Specific Role of svPLA2 in Disordered Coagulation
4.2.2. Role of svMPs in Hemorrhage
4.3. Paralysis
4.3.1. Clinical Features of Paralysis from β-Neurotoxins
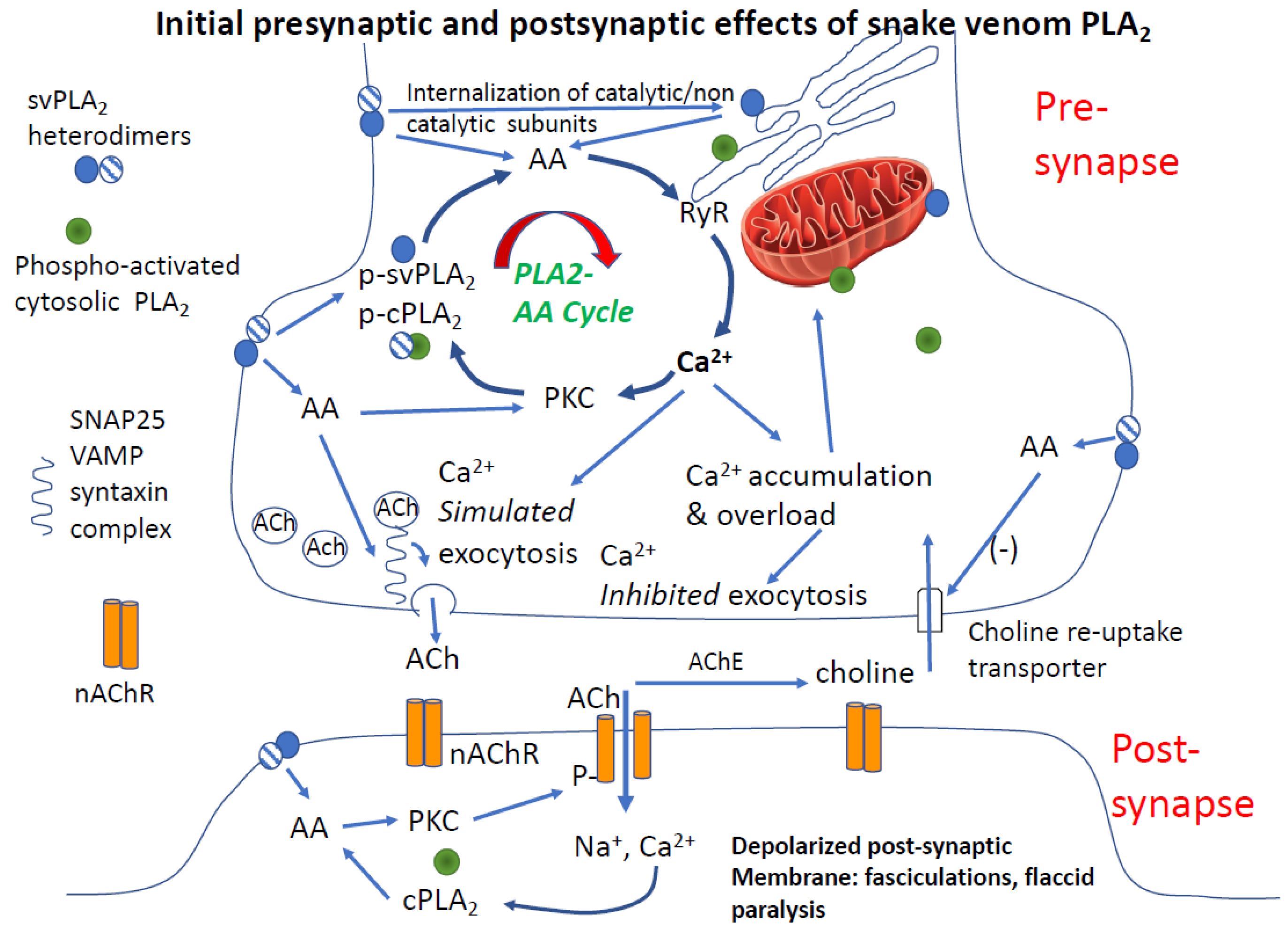
4.3.2. Is PLA2 and Arachidonic Acid Sufficient to Cause Synaptic Failure
4.3.3. Post-Junctional Effects of PLA2 Venoms
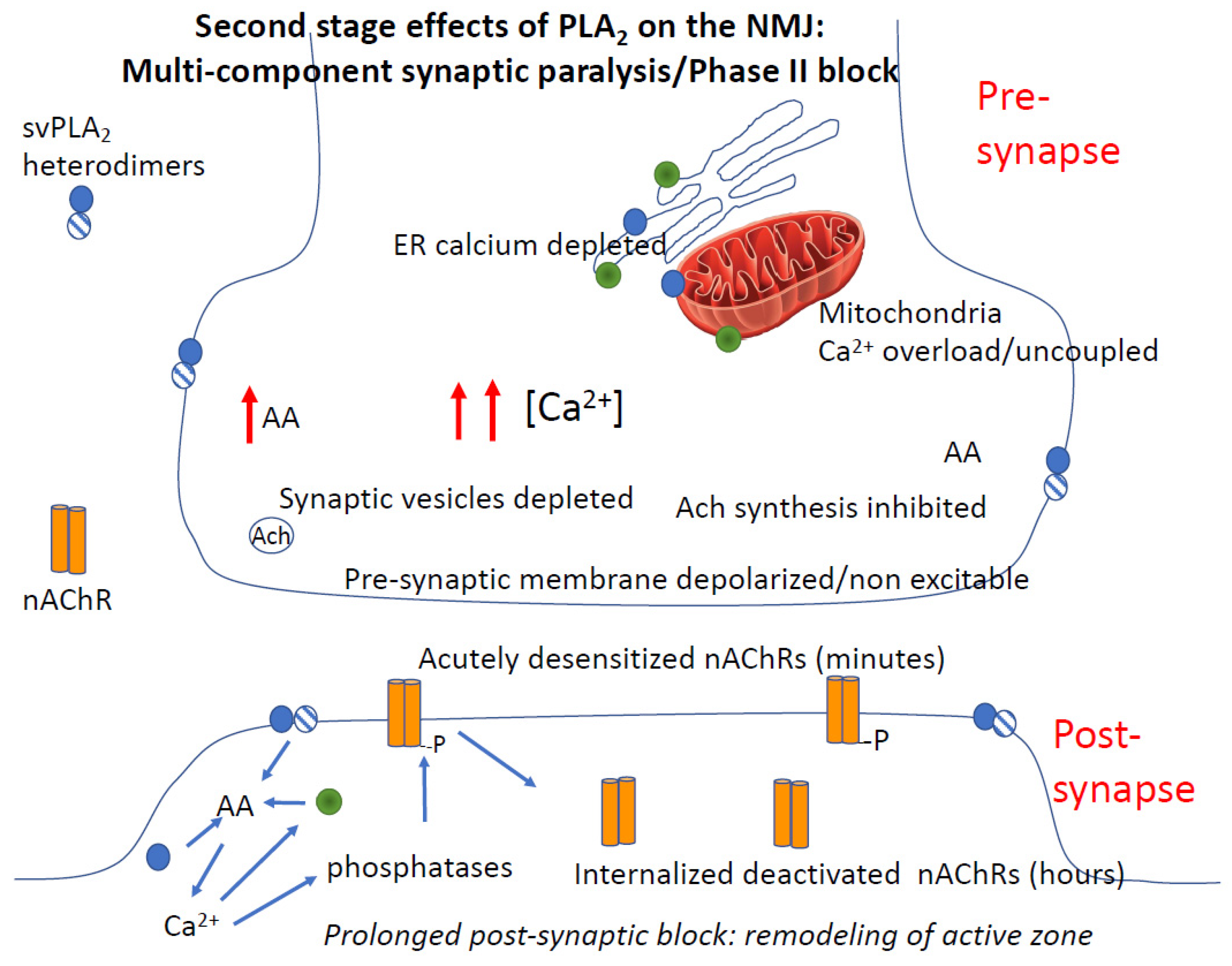
4.3.4. Role of Calcium in PLA2 Mediated Pre- and Post-Junctional Block of Neurotransmission
4.3.5. PLA2 and Rapid Degeneration of the Synapse
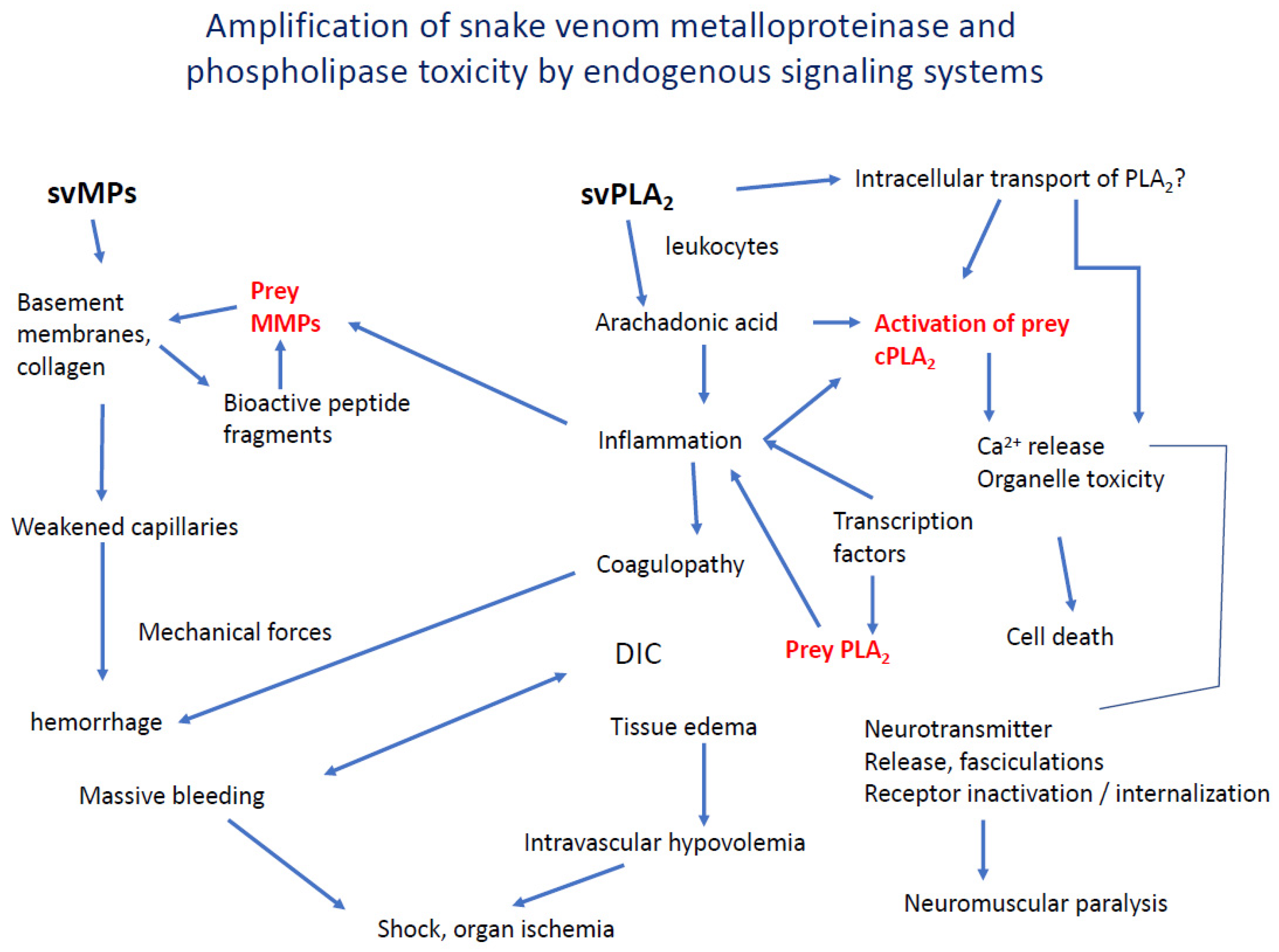
5. Interaction and Amplification of Venom MPs and PLA2s in Inflammation and Coagulation
6. Tools Needed to Dissect the Multi-Compartment Action of Snake Venoms
7. Areas Where New Knowledge Concerning the Cellular Effects of Venom Is Needed
8. Conclusions
Funding
Conflicts of Interest
References
- Longbottom, J.; Shearer, F.M.; Devine, M.; Alcoba, G.; Chappuis, F.; Weiss, D.J.; Ray, S.E.; Ray, N.; Warrell, D.A.; Ruiz de Castaneda, R.; et al. Vulnerability to snakebite envenoming: A global mapping of hotspots. Lancet 2018, 392, 673–684. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17079. [Google Scholar] [CrossRef]
- Isbister, G.K.; Silva, A. Addressing the global problem of snake envenoming. Lancet 2018, 392, 619–620. [Google Scholar] [CrossRef]
- Waiddyanatha, S.; Silva, A.; Siribaddana, S.; Isbister, G.K. Long-term Effects of Snake Envenoming. Toxins 2019, 11, 193. [Google Scholar] [CrossRef]
- Calmette, A. The treatment of anomals poisoned with snakevenom by the injection of antivenomous serum. Br. Med. J. 1896, 2, 399–400. [Google Scholar] [CrossRef]
- Hawgood, B.J. Pioneers of anti-venomous serotherapy: Dr. Vital Brazil. Toxicon 1992, 30, 573–579. [Google Scholar] [CrossRef]
- Lauder Brunton, T.; Fayrer, T. On the nature and physiological action of the poison of Naja tripudians and other Indian venomous snakes. Part I. Proc. R. Soc. 1873, 145, 357–374. [Google Scholar]
- Waheed, H.; Moin, S.F.; Choudhary, M.I. Snake Venom: From Deadly Toxins to Life-saving Therapeutics. Curr. Med. Chem. 2017, 24, 1874–1891. [Google Scholar] [CrossRef]
- Laustsen, A.H. Toxin-centric development approach for next-generation antivenoms. Toxicon 2018, 150, 195–197. [Google Scholar] [CrossRef]
- Tasoulis, T.; Isbister, G.K. A Review and Database of Snake Venom Proteomes. Toxins 2017, 9, 290. [Google Scholar] [CrossRef]
- Cesar, P.H.S.; Braga, M.A.; Trento, M.V.C.; Menaldo, D.L.; Marcussi, S. Snake Venom Disintegrins: An Overview of their Interaction with Integrins. Curr. Drug Targets 2019, 20, 465–477. [Google Scholar] [CrossRef]
- Oliveira-Mendes, B.B.R.; Miranda, S.E.M.; Sales-Medina, D.F.; Magalhaes, B.F.; Kalapothakis, Y.; Souza, R.P.; Cardoso, V.N.; De Barros, A.L.B.; Guerra-Duarte, C.; Kalapothakis, E.; et al. Inhibition of Tityus serrulatus venom hyaluronidase affects venom biodistribution. PLoS Negl. Trop. Dis. 2019, 13, e0007048. [Google Scholar] [CrossRef] [PubMed]
- Kemparaju, K.; Girish, K.S.; Nagaraju, S. Hyaluronidases, a neglected class of glycosidases from snake venom: Beyond a spreading factor. In Handbook of Venoms and Toxins of Reptiles; Mackessey, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2016; Volume 1, pp. 237–261. [Google Scholar]
- Escalante, T.; Rucavado, A.; Fox, J.W.; Gutierrez, J.M. Key events in microvascular damage induced by snake venom hemorrhagic metalloproteinases. J. Proteom. 2011, 74, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Luchini, L.S.G.; Pidde, G.; Squaiella-Baptistao, C.C.; Tambourgi, D.V. Corrigendum: Complement System Inhibition Modulates the Pro-Inflammatory Effects of a Snake Venom Metalloproteinase. Front. Immunol. 2019, 10, 1539. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.F.; Landucci, E.C.; Antunes, E.; Chacur, M.; Cury, Y. Inflammatory effects of snake venom myotoxic phospholipases A2. Toxicon 2003, 42, 947–962. [Google Scholar] [CrossRef]
- Burin, S.M.; Menaldo, D.L.; Sampaio, S.V.; Frantz, F.G.; Castro, F.A. An overview of the immune modulating effects of enzymatic toxins from snake venom. Int J. Biol. Macromol. 2018, 109, 664–671. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Lomonte, B. Phospholipases A2: Unveiling the secrets of a functionally versatile group of snake venom toxins. Toxicon 2013, 62, 27–39. [Google Scholar] [CrossRef]
- Cavalcante, W.L.G.; Noronha-Matos, J.B.; Timoteo, M.A.; Fontes, M.R.M.; Gallacci, M.; Correia-de-Sa, P. Neuromuscular paralysis by the basic phospholipase A2 subunit of crotoxin from Crotalus durissus terrificus snake venom needs its acid chaperone to concurrently inhibit acetylcholine release and produce muscle blockage. Toxicol. Appl. Pharmacol. 2017, 334, 8–17. [Google Scholar] [CrossRef]
- Shier, W.T. Activation of high levels of endogenous phospholipase A2 in cultured cells. Proc. Natl. Acad. Sci. USA 1979, 76, 195–199. [Google Scholar] [CrossRef]
- Katz, B.; Miledi, R. The effect of alpha-bungarotoxin on acetylcholine receptors. Br. J. Pharmacol. 1973, 49, 138–139. [Google Scholar] [CrossRef]
- Lodovicho, M.E.; Costa, T.R.; Bernardes, C.P.; Menaldo, D.L.; Zoccal, K.F.; Carone, S.E.; Rosa, J.C.; Pucca, M.B.; Cerni, F.A.; Arantes, E.C.; et al. Investigating possible biological targets of Bj-CRP, the first cysteine-rich secretory protein (CRISP) isolated from Bothrops jararaca snake venom. Toxicol. Lett. 2017, 265, 156–169. [Google Scholar] [CrossRef]
- Burgers, P.M.; Eckstein, F.; Hunneman, D.H. Stereochemistry of hydrolysis by snake venom phosphodiesterase. J. Biol. Chem. 1979, 254, 7476–7478. [Google Scholar]
- Oguiura, N.; Boni-Mitake, M.; Affonso, R.; Zhang, G. In vitro antibacterial and hemolytic activities of crotamine, a small basic myotoxin from rattlesnake Crotalus durissus. J. Antibiot. 2011, 64, 327–331. [Google Scholar] [CrossRef]
- Ownby, C.L.; Aird, S.D.; Kaiser, I.I. Physiological and immunological properties of small myotoxins from the venom of the midget faded rattlesnake (Crotalus viridis concolor). Toxicon 1988, 26, 319–332. [Google Scholar] [CrossRef]
- Park, M.H.; Jo, M.; Won, D.; Song, H.S.; Han, S.B.; Song, M.J.; Hong, J.T. Snake venom toxin from Vipera lebetina turanica induces apoptosis of colon cancer cells via upregulation of ROS- and JNK-mediated death receptor expression. BMC Cancer 2012, 12, 228. [Google Scholar] [CrossRef]
- Fox, J.W. A brief review of the scientific history of several lesser-known snake venom proteins: L-amino acid oxidases, hyaluronidases and phosphodiesterases. Toxicon 2013, 62, 75–82. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Lohse, B.; Lomonte, B.; Engmark, M.; Gutiérrez, J.M. Selecting key toxins for focused development of elapid snake antivenoms and inhibitors guided by a Toxicity Score. Toxicon. 2015, 104, 43–45. [Google Scholar] [CrossRef]
- Williams, D.J.; Gutiérrez, J.M.; Calvete, J.J.; Wüster, W.; Ratanabanangkoon, K.; Paiva, O.; Brown, N.I.; Casewell, N.R.; Harrison, R.A.; Rowley, P.D.; et al. Ending the drought: New strategies for improving the flow of affordable, effective antivenoms in Asia and Africa. J. Proteom. 2011, 74, 1735–1767. [Google Scholar] [CrossRef]
- Calvete, J.J.; Lomonte, B. A bright future for integrative venomics. Toxicon 2015, 107, 159–162. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Wüster, W.; Cook, D.A.N.; Bolton, F.M.S.; King, S.I.; Pla, D.; Sanz, L.; Calvete, J.J.; Harrison, R.A. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 9205–9210. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Lomonte, B.; Lohse, B.; Fernández, J.; Gutiérrez, J.M. Unveiling the nature of black mamba (Dendroaspis polylepis) venom through venomics and antivenom immunoprofiling: Identification of key toxin targets for antivenom development. J. Proteom. 2015, 119, 126–142. [Google Scholar] [CrossRef]
- Bordon, K.C.; Perino, M.G.; Giglio, J.R.; Arantes, E.C. Isolation, enzymatic characterization and antiedematogenic activity of the first reported rattlesnake hyaluronidase from Crotalus durissus terrificus venom. Biochimie 2012, 94, 2740–2748. [Google Scholar] [CrossRef]
- Slotta, K.H.; Fraenkel-Conrat, H. Two active proteins from rattlesnake venom. Nature 1938, 142, 213. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Barnard, E.A.; Chiu, T.H.; Lapa, A.J.; Dolly, J.O.; Jansson, S.E.; Daly, J.; Witkop, B. Acetylcholine receptor and ion conductance modulator sites at the murine neuromuscular junction: Evidence from specific toxin reactions. Proc. Natl. Acad. Sci. USA 1973, 70, 949–953. [Google Scholar] [CrossRef]
- Sousa, I.D.L.; Barbosa, A.R.; Salvador, G.H.M.; Frihling, B.E.F.; Santa-Rita, P.H.; Soares, A.M.; Pessoa, H.L.F.; Marchi-Salvador, D.P. Secondary hemostasis studies of crude venom and isolated proteins from the snake Crotalus durissus terrificus. Int. J. Biol. Macromol. 2019, 131, 127–133. [Google Scholar] [CrossRef]
- Jorge, M.T.; Ribeiro, L.A. Epidemiology and clinical features of South American rattlesnakes (Crotalus durissus) envenomation. Rev. Do Inst. Demedicina Trop. De São Paulo 1992, 34, 347–354. [Google Scholar] [CrossRef]
- Silveira, P.V.; Nishioka Sde, A. South American rattlesnake bite in a Brazilian teaching hospital. Clinical and epidemiological study of 87 cases, with analysis of factors predictive of renal failure. Trans. R Soc. Trop. Med. Hyg. 1992, 86, 562–564. [Google Scholar] [CrossRef]
- Oliveira, I.S.; Cardoso, I.A.; Bordon, K.D. Global proteomic and functional analysis of Crotalus durissus collilineatus individual venom variation and its impact on envenoming. J. Proteom. 2019, 191, 153–165. [Google Scholar] [CrossRef]
- Sanhajariya, S.; Duffull, S.B.; Isbister, G.K. Pharmacokinetics of snake venom. Toxins 2018, 10, 73. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Escalante, T.; Rucavado, A.; Herrera, C.; Fox, J.W. A Comprehensive View of the Structural and Functional Alterations of Extracellular Matrix by Snake Venom Metalloproteinases (SVMPs): Novel Perspectives on the Pathophysiology of Envenoming. Toxins 2016, 8, 304. [Google Scholar] [CrossRef]
- Reynolds, J.J. Collagenases and tissue inhibitors of metalloproteinases: A functional balance in tissue degradation. Oral Dis. 1996, 2, 70–76. [Google Scholar] [CrossRef]
- Diraviyam, K.; Murray, D. Computational analysis of the membrane association of group IIA secreted phospholipases A2: A differential role for electrostatics. Biochemistry 2006, 45, 2584–2598. [Google Scholar] [CrossRef]
- Oberg, S.G.; Kelly, R.B. Saturable binding to cell membranes of the presynaptic neurotoxin β-bungarotoxin. Biochem. Biophys. Acta 1976, 433, 662–673. [Google Scholar] [CrossRef]
- MacDermot, J.W.; Westgaard, R.H.; Thompson, E.J. The binding of [3H]-pyridoxylated Beta-bungarotoxin to a high molecular weight protein receptor. Biochem. J. 1978, 175, 282–288. [Google Scholar] [CrossRef]
- Harris, J.B.; Scott-Davey, T. Secreted Phospholipases A2 of Snake Venoms: Effects on the Peripheral Neuromuscular System with Comments on the Role of Phospholipases A2 in Disorders of the CNS and Their Uses in Industry. Toxins 2013, 5, 2533–2571. [Google Scholar] [CrossRef]
- Lambeau, G.; Schmid-Alliana, A.; Lazdunski, M.; Barhanin, J. Identification and purification of a very high affinity binding protein for toxic phospholipases A2 in skeletal muscle. J. Biol. Chem. 1990, 265, 9526–9532. [Google Scholar]
- Nakashima, K.; Ogawa, T.; Oda, N.; Hattori, M.; Sakaki, Y.; Kihara, H.; Ohno, M. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isozymes. Proc. Natl. Acad. Sci. USA 1993, 90, 5964–5968. [Google Scholar] [CrossRef]
- Kini, R.M.; Evans, H.J. A model to explain the pharmacological effects of snake venom phospholipases A2. Toxicon 1989, 27, 613–635. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Logonder, U.; Jenko-Praznikar, Z.; Scott-Davey, T.; Pungercar, J.; Krizaj, I.; Harris, J.B. Ultrastructural evidence for the uptake of a neurotoxic snake venom phospholipase A2 into mammalian motor nerve terminals. Exp. Neurol. 2009, 219, 591–594. [Google Scholar] [CrossRef]
- Rigoni, M.; Paoli, M.; Milanesi, E.; Caccin, P.; Rasola, A.; Bernardi, P.; Montecucco, C. Snake phospholipase A2 neurotoxins enter neurons, bind specifically to mitochondria, and open their transition pores. J. Biol. Chem. 2008, 283, 34013–34020. [Google Scholar] [CrossRef] [PubMed]
- Spolaore, B.; Fernandez, J.; Lomonte, B.; Massimino, M.L.; Tonello, F. Enzymatic labelling of snake venom phospholipase A2 toxins. Toxicon 2019, 170, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Sribar, J.; Oberckal, J.; Krizaj, I. Understanding the molecular mechanism underlying the presynaptic toxicity of secreted phospholipases A(2): An update. Toxicon 2014, 89, 9–16. [Google Scholar] [CrossRef]
- Borges, R.J.; Lemke, N.; Fontes, M.R.M. PLA2-like proteins myotoxic mechanism: A dynamic model description. Sci. Rep. 2017, 7, 15514. [Google Scholar] [CrossRef]
- Leiguez, E.; Giannotti, K.C.; Moreira, V.; Matsubara, M.H.; Gutiérrez, J.M.; Lomonte, B.; Rodríguez, J.P.; Balsinde, J.; Teixeira, C. Critical role of TLR2 and MyD88 for functional response of macrophages to a group IIA-secreted phospholipase A2 from snake venom. PLoS ONE 2014, 9, e93741. [Google Scholar] [CrossRef]
- Martin, S.A.; Brash, A.R.; Murphy, R.C. The discovery and early structural studies of arachidonic acid. J. Lipid Res. 2016, 57, 1126–1132. [Google Scholar] [CrossRef]
- Choumet, V.; Saliou, B.; Fideler, L.; Chen, Y.C.; Gubensek, F.; Bon, C.; Delot, E. Snake-venom phospholipase A2 neurotoxins. Potentiation of a single-chain neurotoxin by the chaperon subunit of a two-component neurotoxin. Eur. J. Biochem. 1993, 211, 57–62. [Google Scholar] [CrossRef]
- Doley, R.; Kini, R.M. Protein complexes in snake venom. Cell. Mol. Life Sci. 2009, 66, 2851–2871. [Google Scholar] [CrossRef]
- Laustsen, A.H. Toxin synergism in snake venoms. Toxin Rev. 2016, 35, 165–170. [Google Scholar] [CrossRef]
- Salvador, G.H.M.; Cardoso, F.F.; Gomes, A.A.; Cavalcante, W.L.G.; Gallacci, M.; Fontes, M.R.M. Search for efficient inhibitors of myotoxic activity induced by ophidian phospholipase A2-like proteins using functional, structural and bioinformatics approaches. Sci. Rep. 2019, 9, 510. [Google Scholar] [CrossRef]
- Pla, D.; Sanz, L.; Quesada-Bernat, S.; Villalta, M.; Baal, J.; Chowdhury, M.A.W.; Leon, G.; Gutierrez, J.M.; Kuch, U.; Calvete, J.J. Phylovenomics of Daboia russelii across the Indian subcontinent. Bioactivities and comparative in vivo neutralization and in vitro third-generation antivenomics of antivenoms against venoms from India, Bangladesh and Sri Lanka. J. Proteom. 2019, 207, 103443. [Google Scholar] [CrossRef]
- Paillamanque, J.; Sanchez-Tusie, A.; Carmona, E.M.; Trevino, C.L.; Sandoval, C.; Nualart, F.; Osses, N.; Reyes, J.G. Arachidonic acid triggers [Ca2+] i increases in rat round spermatids by a likely GPR activation, ERK signalling and ER/acidic compartments Ca2+ release. PLoS ONE 2017, 12, e0172128. [Google Scholar] [CrossRef]
- Williams, D.J.; Faiz, M.A.; Abela-Ridder, B.; Ainsworth, S.; Bulfone, T.C.; Nickerson, A.D.; Habib, A.G.; Junghanss, T.; Fan, H.W.; Turner, M.; et al. Strategy for a globally coordinated response to a priority neglected tropical disease: Snakebite envenoming. PLoS Negl. Trop. Dis. 2019, 13, e0007059. [Google Scholar] [CrossRef]
- Bulfone, T.C.; Samuel, S.P.; Bickler, P.E.; Lewin, M.R. Developing Small Molecule Therapeutics for the Initial and Adjunctive Treatment of Snakebite. J. Trop. Med. 2018, 2018, 4320175. [Google Scholar] [CrossRef]
- Herzel, B.J.; Samuel, S.P.; Bulfone, T.C.; Raj, C.S.; Lewin, M.; Kahn, J.G. Snakebite: An Exploratory Cost-Effectiveness Analysis of Adjunct Treatment Strategies. Am. J. Trop. Med. Hyg. 2018, 99, 404–412. [Google Scholar] [CrossRef]
- Lewin, M.R.; Gilliam, L.L.; Gilliam, J.; Samuel, S.P.; Bulfone, T.C.; Bickler, P.E.; Gutierrez, J.M. Delayed LY333013 (Oral) and LY315920 (Intravenous) Reverse Severe Neurotoxicity and Rescue Juvenile Pigs from Lethal Doses of Micrurus fulvius (Eastern Coral Snake) Venom. Toxins 2018, 10, 479. [Google Scholar] [CrossRef]
- Lewin, M.R.; Gutierrez, J.M.; Samuel, S.P.; Herrera, M.; Bryan-Quiros, W.; Lomonte, B.; Bickler, P.E.; Bulfone, T.C.; Williams, D.J. Delayed Oral LY333013 Rescues Mice from Highly Neurotoxic, Lethal Doses of Papuan Taipan (Oxyuranus scutellatus) Venom. Toxins 2018, 10, 380. [Google Scholar] [CrossRef]
- Salvador, G.; Gomes, A.A.S.; Bryan-Quirós, W.; Fernández, J.; Lewin, M.R.; Gutiérrez, J.M.; Lomonte, B.; Fonte, M.R.M. Structural basis for phospholipase A2-like toxin inhibition by the synthetic compound Varespladib (LY315920). Nat. Sci. Rep. 2019, in press. [Google Scholar] [CrossRef]
- Nagase, H. Metalloproteases. Curr. Protoc. Protein Sci. 2001, 24, 21.4.1–21.4.13. [Google Scholar] [CrossRef]
- Saklatvala, J.; Dean, J.; Clark, A. Control of the expression of inflammatory response genes. Biochem. Soc. Symp. 2003, 70, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Papavassiliou, A.G. Molecular mechanisms regulating matrix metalloproteinases. Curr. Top. Med. Chem. 2012, 12, 1095–1112. [Google Scholar] [CrossRef] [PubMed]
- Ownby, C.L.; Bjarnason, J.; Tu, A.T. Hemorrhagic toxins from rattlesnake (Crotalus atrox) venom. Pathogenesis of hemorrhage induced by three purified toxins. Am. J. Pathol. 1978, 93, 201–218. [Google Scholar] [PubMed]
- McKay, D.G.; Moroz, C.; DeVries, A.; Csavossy, I.; Cruse, V. The action of hemorrhagin and phospholipase derived from Vipera palestinae venom on the microcirculation. Lab. Intestig. 1970, 22, 387–399. [Google Scholar]
- Zelanis, A.; Oliveira, A.K.; Prudova, A.; Huesgen, P.F.; Tashima, A.K.; Kizhakkedathu, J.; Overall, C.M.; Serrano, S.M.T. Deep Profiling of the Cleavage Specificity and Human Substrates of Snake Venom Metalloprotease HF3 by Proteomic Identification of Cleavage Site Specificity (PICS) Using Proteome Derived Peptide Libraries and Terminal Amine Isotopic Labeling of Substrates (TAILS) N-Terminomics. J. Proteome Res. 2019, 18, 3419–3428. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.M.; Rucavado, A.; Escalante, T.; Herrera, C.; Fernandez, J.; Lomonte, B.; Fox, J.W. Unresolved issues in the understanding of the pathogenesis of local tissue damage induced by snake venoms. Toxicon 2018, 148, 123–131. [Google Scholar] [CrossRef]
- Rucavado, A.; Lomonte, B.; Ovadia, M.; Gutierrez, J.M. Local tissue damage induced by BaP1, a metalloproteinase isolated from Bothrops asper (Terciopelo) snake venom. Exp. Mol. Pathol. 1995, 63, 186–199. [Google Scholar] [CrossRef]
- Fernandes, C.M.; Pereira Teixeira Cde, F.; Leite, A.C.; Gutiérrez, J.M.; Rocha, F.A. The snake venom metalloproteinase BaP1 induces joint hypernociception through TNF-alpha and PGE2-dependent mechanisms. Br. J. Pharmacol. 2007, 151, 1254–1261. [Google Scholar] [CrossRef]
- Fernandes, C.M.; Zamuner, S.R.; Zuliani, J.P.; Rucavado, A.; Gutierrez, F.M.; Teixeira, F. Inflammatory effects of BaP1 a metalloproteinase isolated from Bothrops asper snake venom: Leukocyte recruitment and release of cytokines. Toxicon 2006, 47, 549–559. [Google Scholar] [CrossRef]
- Fieber, C.; Baumann, P.; Vallon, R.; Termeer, C.; Simon, J.C.; Hofmann, M.; Angel, P.; Herrlich, P.; Sleeman, J.P. Hyaluronan-oligosaccharide-induced transcription of metalloproteases. J. Cell Sci. 2004, 117, 359–367. [Google Scholar] [CrossRef]
- Sivaramakrishnan, V.; Ilamathi, M.; Girish, K.S.; Kemparaju, K.; Rangappa, K.S.; Dhananjaya, B.L. Viper venom hyaluronidase and its potential inhibitor analysis: A multipronged computational investigation. J. Biomol. Struct. Dyn. 2017, 35, 1979–1989. [Google Scholar] [CrossRef]
- Lauridsen, L.P.; Laustsen, A.H.; Lomonte, B.; Gutiérrez, J.M. Toxicovenomics and antivenom profiling of the Eastern green mamba snake (Dendroaspis angusticeps). J. Proteom. 2016, 136, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Pla, D.; Pérez, A.; Rodríguez, Y.; Zavaleta, A.; Salas, M.; Lomonte, B.; Calvete, J.J. Venomic Analysis of the Poorly Studied Desert Coral Snake, Micrurus tschudii tschudii, Supports the 3FTx/PLA₂ Dichotomy across Micrurus Venoms. Toxins 2016, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, L.O.; Kazandjian, T.; Slagboom, J.; Bruyneel, B.; Ainsworth, S.; Alsolaiss, J.; Wagstaff, S.C.; Whiteley, G.; Harrison, R.A.; Ulens, C.; et al. A Decoy-Receptor Approach Using Nicotinic Acetylcholine Receptor Mimics Reveals Their Potential as Novel Therapeutics Against Neurotoxic Snakebite. Front. Pharmacol. 2019, 10, 848. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Starkl, P.; Marichal, T.; Tsai, M. Mast cells and IgE can enhance survival during innate and acquired host responses to venoms. Trans. Am. Clin. Clim. Assoc. 2017, 128–221, 193–221. [Google Scholar]
- Stone, S.F.; Isbister, G.K.; Shahmy, S.; Mohamed, F.; Abeysinghe, C.; Karunathilake, H.; Ariaratnam, A.; Jacoby-Alner, T.E.; Cotterell, C.L.; Brown, S.G. Immune response to snake envenoming and treatment with antivenom; complement activation, cytokine production and mast cell degranulation. PLoS Negl. Trop. Dis. 2013, 7, e2326. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Fernandes, C.M.; Leiguez, E.; Chudzinski-Tavassi, A.M. Inflammation Induced by Platelet-Activating Viperid Snake Venoms: Perspectives on Thromboinflammation. Front. Immunol. 2019, 10, 2082. [Google Scholar] [CrossRef]
- Wang, S.Z.; Qin, Z.H. Anti-Inflammatory and Immune Regulatory Actions of Naja naja atra Venom. Toxins 2018, 10, 100. [Google Scholar] [CrossRef]
- Silva-de-Franca, F.; Villas-Boas, I.M.; Serrano, S.M.T.; Cogliati, B.; Chudzinski, S.A.A.; Lopes, P.H.; Kitano, E.S.; Okamoto, C.K.; Tambourgi, D.V. Naja annulifera Snake: New insights into the venom components and pathogenesis of envenomation. PLoS Negl. Trop. Dis. 2019, 13, e0007017. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Rucavado, A.; Chaves, F.; Diaz, C.; Escalante, T. Experimental pathology of local tissue damage induced by Bothrops asper snake venom. Toxicon 2009, 54, 958–975. [Google Scholar] [CrossRef]
- Teixeira, C.; Cury, Y.; Moreira, V.; Picolo, G.; Chaves, F. Inflammation induced by Bothrops asper venom. Toxicon 2009, 54, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Damerau, B.; Lege, L.; Oldigs, H.D.; Vogt, W. Histamine release, formation of prostaglandin-like activity (SRS-C) and mast cell degranulation by the direct lytic factor (DLF) and phospholipase A of cobra venom. Naunyn Schmiedebergs Arch. Pharmacol. 1975, 287, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Brain, S.; Lewis, G.P.; Whittle, B.J. Actions of phospholipase-A on mast-cell histamine release and paw oedema in the rat [proceedings]. Br. J. Pharmacol. 1977, 59, 440P–441P. [Google Scholar] [PubMed]
- Boeno, C.N.; Paloschi, M.V.; Lopes, J.A.; Pires, W.L.; Setubal, S.D.S.; Evangelista, J.R.; Soares, A.M.; Zuliani, J.P. Inflammasome Activation Induced by a Snake Venom Lys49-Phospholipase A2 Homologue. Toxins 2019, 12, 22. [Google Scholar] [CrossRef]
- Bhowmick, R.; Clark, S.; Bonventre, J.V.; Leong, J.M.; McCormick, B.A. Cytosolic Phospholipase A2α Promotes Pulmonary Inflammation and Systemic Disease during Streptococcus pneumoniae Infection. Infect. Immun. 2017, 85, e00280-17. [Google Scholar] [CrossRef]
- Palangasinghe, D.R.; Weerakkody, R.M.; Dalpatadu, C.G.; Gnanathasan, C.A. A fatal outcome due to pulmonary hemorrhage following Russell’s viper bite. Saudi Med. J. 2015, 36, 634–637. [Google Scholar] [CrossRef]
- Gnanathasan, A.; Rodrigo, C. Pulmonary effects and complications of snakebites. Chest 2014, 146, 1403–1412. [Google Scholar] [CrossRef]
- Siddall, E.; Khatri, M.; Radhakrishnan, J. Capillary leak syndrome: Etiologies, pathophysiology, and management. Kidney Int. 2017, 92, 37–46. [Google Scholar] [CrossRef]
- Agarwal, R.; Singh, A.P.; Aggarwal, A.N. Pulmonary oedema complicating snake bite due to Bungarus caeruleus. Singapore Med. J. 2007, 48, e227–e230. [Google Scholar] [PubMed]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Bittenbinder, M.A.; Zdenek, C.N.; Op den Brouw, B.; Youngman, N.J.; Dobson, J.S.; Naude, A.; Vonk, F.J.; Fry, B.G. Coagulotoxic Cobras: Clinical Implications of Strong Anticoagulant Actions of African Spitting Naja Venoms That Are Not Neutralised by Antivenom but Are by LY315920 (Varespladib). Toxins 2018, 10, 516. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.C.; Sundaram, M.S.; Mohan, C.D.; Rangappa, S.; Bulusu, K.C.; Fuchs, J.E.; Girish, K.S.; Bender, A.; Basappa; Rangappa, K.S. A One Pot Synthesis of Novel Bioactive Tri-Substitute-Condensed-Imidazopyridines that Targets Snake Venom Phospholipase A2. PLoS ONE 2015, 10, e0131896. [Google Scholar] [CrossRef] [PubMed]
- Ranawaka, U.K.; Lalloo, D.G.; De Silva, H. Neurotoxicity in snakebite—The limits of our knowledge. PLoS Negl. Trop. Dis. 2013, 7, e2302. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.C. Neuromuscular block. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S277–S286. [Google Scholar] [CrossRef]
- Bamford, N.J.; Sprinkle, S.B.; Cudmore, L.A.; Cullimore, A.M.; Van Eps, A.W.; Verdegaal, E.; Tennent-Brown, B.S. Elapid snake envenomation in horses: 52 cases (2006-2016). Equine Vet. J. 2018, 50, 196–201. [Google Scholar] [CrossRef]
- Bosak, A.R.; Ruha, A.M.; Graeme, K.A. A case of neurotoxicity following envenomation by the Sidewinder rattlesnake, Crotalus cerastes. J. Med. Toxicol. 2014, 10, 229–231. [Google Scholar] [CrossRef]
- Richardson, W.H.; Goto, C.S.; Gutglass, D.J.; Williams, S.R.; Clark, R.F. Rattlesnake envenomation with neurotoxicity refractory to treatment with crotaline Fab antivenom. Clin. Toxicol. (Phila.) 2007, 45, 472–475. [Google Scholar] [CrossRef]
- Katz, B.; Thesleff, S. A study of the desensitization produced by acetylcholine at the motor end-plate. J. Physiol. 1957, 29, 63–80. [Google Scholar] [CrossRef]
- Khaziev, E.; Samigullin, D.; Zhilyakov, N.; Fatikhov, N.; Bukharaeva, E.; Verkhratsky, A.; Nikolsky, E. Acetylcholine-Induced Inhibition of Presynaptic Calcium Signals and Transmitter Release in the Frog Neuromuscular Junction. Front. Physiol. 2016, 7, 621. [Google Scholar] [CrossRef]
- Eilers, H.; Schaeffer, E.; Bickler, P.E.; Forsayeth, J.R. Functional deactivation of the major neuronal nicotinic receptor caused by nicotine and a protein kinase C-dependent mechanism. Mol. Pharmacol. 1997, 52, 1105–1112. [Google Scholar] [CrossRef]
- Vulfius, C.A.; Kasheverov, I.E.; Kryukova, E.V.; Spirova, E.N.; Shelukhina, I.V.; Starkov, V.G.; Andreeva, T.V.; Faure, G.; Zouridakis, M.; Tsetlin, V.I.; et al. Pancreatic and snake venom presynaptically active phospholipases A2 inhibit nicotinic acetylcholine receptors. PLoS ONE 2017, 12, e0186206. [Google Scholar] [CrossRef] [PubMed]
- Bon, C. Synergism of the two subunits of crotoxin. Toxicon 1982, 20, 105–109. [Google Scholar] [CrossRef]
- Boksa, P.; Mykita, S.; Collier, B. Arachidonic acid inhibits choline uptake and depletes acetylcholine content in rat cerebral cortical synaptosomes. J. Neurochem. 1988, 50, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chen, T.F.; Lee, C.Y. Studies of the presynaptic effect of -bungarotoxin on neuromuscular transmission. J. Pharmacol. Exp. 1973, 184, 339–345. [Google Scholar]
- Garcia-Martinez, V.; Gimenez-Molina, Y.; Villanueva, J.; Darios, F.D.; Davletov, B.; Gutierrez, L.M. Emerging evidence for the modulation of exocytosis by signalling lipids. FEBS Lett. 2018, 592, 3493–3503. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca(2+) Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef]
- Schaechter, J.D.; Benowitz, L.I. Activation of protein kinase C by arachidonic acid selectively enhances the phosphorylation of GAP-43 in nerve terminal membranes. J. Neurosci. 1993, 13, 4361–4371. [Google Scholar] [CrossRef]
- St John, P.A.; Gordon, H. Agonists cause endocytosis of nicotinic acetylcholine receptors on cultured myotubes. J. Neurobiol. 2001, 49, 212–223. [Google Scholar] [CrossRef]
- Tedesco, E.; Rigoni, M.; Caccin, P.; Grishin, E.; Rossetto, O.; Montecucco, C. Calcium overload in nerve terminals of cultured neurons intoxicated by alpha-latrotoxin and snake PLA2 neurotoxins. Toxicon 2009, 54, 138–144. [Google Scholar] [CrossRef]
- Cintra-Francischinelli, M.; Pizzo, P.; Angulo, Y.; Gutierrez, J.M.; Montecucco, C.; Lomonte, B. The C-terminal region of a Lys49 myotoxin mediates Ca2+ influx in C2C12 myotubes. Toxicon 2010, 55, 590–596. [Google Scholar] [CrossRef]
- Angulo, Y.; Lomonte, B. Differential susceptibility of C2C12 myoblasts and myotubes to group II phospholipase A2 myotoxins from crotalid snake venoms. Cell Biochem. Funct. 2005, 23, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Angulo, Y.; Rufini, S.; Cho, W.; Giglio, J.R.; Ohno, M.; Daniele, J.J.; Geoghegan, P.; Gutierrez, J.M. Comparative study of the cytolytic activity of myotoxic phospholipases A2 on mouse endothelial (tEnd) and skeletal muscle (C2C12) cells in vitro. Toxicon 1999, 37, 145–158. [Google Scholar] [CrossRef]
- Bickler, P.E.; Fahlman, C.S.; Gray, J.; McKleroy, W. Inositol 1,4,5-triphosphate receptors and NAD(P)H mediate Ca2+ signaling required for hypoxic preconditioning of hippocampal neurons. Neuroscience 2009, 160, 51–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Verma, M.; Wills, Z.; Chu, C.T. Excitatory Dendritic Mitochondrial Calcium Toxicity: Implications for Parkinson’s and Other Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 523. [Google Scholar] [CrossRef]
- Harris, J.B.; Grubb, B.D.; Maltin, C.A.; Dixon, R. The neurotoxicity of the venom phospholipases A(2), notexin and taipoxin. Exp. Neurol. 2000, 161, 517–526. [Google Scholar] [CrossRef]
- Dixon, R.W.; Harris, J.B. Nerve terminal damage by b-bungarotoxin: it’s clinical significance. Am. J. Pathol. 1999, 154, 447–455. [Google Scholar] [CrossRef]
- Chibalin, A.V.; Benziane, B.; Zakyrjanova, G.F.; Kravtsova, V.V.; Krivoi, I.I. Early endplate remodeling and skeletal muscle signaling events following rat hindlimb suspension. J. Cell Physiol. 2018, 233, 6329–6336. [Google Scholar] [CrossRef]
- Bohme, M.A.; McCarthy, A.W.; Grasskamp, A.T.; Beuschel, C.B.; Goel, P.; Jusyte, M.; Laber, D.; Huang, S.; Rey, U.; Petzoldt, A.G.; et al. Rapid active zone remodeling consolidates presynaptic potentiation. Nat. Commun. 2019, 10, 1085. [Google Scholar] [CrossRef]
- Deem, S. Intensive-care-unit-acquired muscle weakness. Respir. Care 2006, 51, 1042–1052. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Venom Component | Primary Pathologic Effect | Site of Action | Timing of Effect | Enzymatic or Non-Enzymatic | Venom Action Amplified by Prey |
|---|---|---|---|---|---|
| Disintegrins | Inhibit cell-ECM, loosen anchoring tissue [10] | Interstitial spaces | Intermediate and late? | Non | Yes, augments inflammation [11] |
| Hyaluronidases | Loosens tissue, enhances venom spread [12], exposes tissue factor | Capillaries and Interstitial spaces | Intermediate | Enzymatic | Yes, augments inflammation, coagulopathy [13] |
| Metalloproteases | Loosens/digests basal lamina [14] | Capillaries connective tissue | Intermediate | Enzymatic | Yes, bioactive peptides [15], gene expression |
| Serine Proteases | Inhibit coagulation, anti-thrombin effect [8] | Blood | Rapid | Enzymatic | Yes, signal cascades |
| Antithrombins | Hydrolysis of thrombin, clot destabilizer [8] | Blood | Rapid | Enzymatic | Unknown |
| PLA2s (inflammation, coagulation) | Production of arachidonic acid, mediators of inflammation [16,17] | leukocytes, platelets, endothelial cells | Rapid | Enzymatic and non-enzymatic subunits [18] | Yes, Ca2+, arachidonate, phosphorylation, gene expr. [17] |
| PLA2s (-neurotoxin) | Paralysis | Neuromuscular junction [19] | Usually rapid but may evolve slowly | Enzymatic and non-enzymatic | Yes, homologous protein activation [20] |
| 3FTx (-neurotoxins) | Paralysis/anticholinergic | Antagonists of nicotinic/muscarinic receptors [21] | Rapid | Direct | No |
| Cysteine-rich secretory proteins (CRISPS) | Target ion channels, Ca2+ release | Endothelium, leukocytes [22] | Delayed | unknown | Unknown |
| Kallikrein-like proteins | Shock, physiological disturbance [8] | Vasodilator | Rapid | Direct | Yes, amplifies inflammation |
| Phosphodiesterases | Hydrolysis of cyclic nucleotides/cell signaling/vasodilation [23] | Cell membrane, intracellular | Intermediate | Enzymatic | Yes, cell signaling pathways |
| Myotoxins | Cell damage [24,25] | Sarcolemma | Intermediate Delayed | Direct | Possible, overlap/identity to some PLA2s |
| Activators of cell death receptors DR4 and DR5 | Programmed cell death (apoptosis) [26] | Liver, kidneys, muscle | Delayed | Direct | Yes, cell apoptosis machinery |
| L-Amino Acid Oxidases | Free radicals tissue damage, immune activation [27] | Blood, extracellular fluid | Intermediate | Enzymatic | Yes, cytokine gene expression |
| Key Question/Hypothesis | Best Evidence for | Evidence Against | What Is Needed? |
|---|---|---|---|
| Is the catalytic action of svPLA2 responsible for paralysis? | Fasciculations [107], Pre-junctional effects, Ca2+ measurements [54], arachidonic acid | EM of altered NMJ morphology Post junctional effects [127] | Electrophysiology calcium imaging knockout mice. Small molecule inhibitor studies |
| Is endogenous PLA2 activation required for paralysis by β-toxins? | Small molecule inhibitors of endogenous PLA2 reverse paralysis [67,68] | None, excluding possible α-toxin effects of some PLA2s [113] | Ca2+-chelators, ER calcium imaging/release inhibitors should block synaptic failure |
| Do β-toxins cause reversible changes in the NMJ? | Delayed rescue by SMTs possible [67,68] | Ultrastructural images of damage [127] | Small molecule inhibitor studies, longer term assessment of NMJ structure |
| Is recruitment of prey PLA2 required to initiate or sustain inflammation? | Inflammation is sustained for long duration, failure of serotherapy to address [4] | No specific evidence against | Small molecule inhibitors selective for endogenous PLA2, cytokine gene arrays, genetic models |
| Can inhibition of endogenous PLA2 prevent organ toxicity? | Not studied | Organ damage caused by non PLA2 venom components [8] | Assessment of renal, hepatic, pulmonary function. Small molecule inhibitor studies |
| Are svPLA2s and svMPs synergistic in producing inflammation? | Elevations of cytokines, expression of MMPs [17] | Evidence lacking | Cytokine gene arrays with and without SMTs Cytokine levels |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bickler, P.E. Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways. Toxins 2020, 12, 68. https://doi.org/10.3390/toxins12020068
Bickler PE. Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways. Toxins. 2020; 12(2):68. https://doi.org/10.3390/toxins12020068
Chicago/Turabian StyleBickler, Philip E. 2020. "Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways" Toxins 12, no. 2: 68. https://doi.org/10.3390/toxins12020068
APA StyleBickler, P. E. (2020). Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways. Toxins, 12(2), 68. https://doi.org/10.3390/toxins12020068