Water Thermodynamics of Peptide Toxin Binding Sites on Ion Channels


Abstract
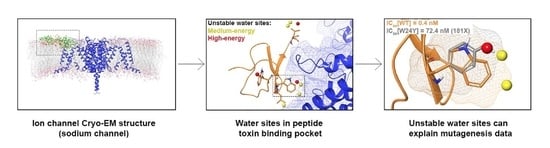
1. Introduction
2. Results
2.1. Apo WaterMaps of Peptide Toxin Binding Sites on Ion Channels
2.2. Frequency Analysis of Residues That Overlap Unstable Waters
2.3. Holo WaterMap of ProTx2 Bbound to a Nav Channel
3. Discussion
3.1. Unstable Water Sites Are Present in the Peptide Toxin Binding Sites of Ion Channels
3.2. Unstable Water Sites Overlap With Most, but Not All, Residues Identified by Mutagenesis Efforts as Functionally Important
3.3. Additional Unstable Water Sites Exist on the Nav Channel Near ProTx2 and Could Be Targeted by Virtual and Experimental Screening
4. Materials and Methods
4.1. Protein Preparation
4.2. WaterMap Calculations
4.3. WaterMap Analysis
4.4. Plotting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holford, M.; Daly, M.; King, G.F.; Norton, R.S. Venoms to the rescue. Science 2018, 361, 842–844. [Google Scholar] [CrossRef]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Kasai, M.; Lee, C.-Y. Use of a Snake Venom Toxin to Characterize the Cholinergic Receptor Protein. Proc. Natl. Acad. Sci. USA 1970, 67, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.J.; Sanjakdar, S.S.; Muldoon, P.P.; McIntosh, J.M.; Damaj, M.I. The α3β4* nicotinic acetylcholine receptor subtype mediates nicotine reward and physical nicotine withdrawal signs independently of the α5 subunit in the mouse. Neuropharmacology 2013, 70, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nat. Cell Biol. 2004, 430, 232–235. [Google Scholar] [CrossRef]
- Pennington, M.W.; Czerwinski, A.; Norton, R.S. Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [Google Scholar] [CrossRef]
- Neff, R.A.; Flinspach, M.; Gibbs, A.; Shih, A.Y.; Minassian, N.A.; Liu, Y.; Fellows, R.; Libiger, O.; Young, S.; Pennington, M.W.; et al. Comprehensive engineering of the tarantula venom peptide huwentoxin-IV to inhibit the human voltage-gated sodium channel hNav1.7. J. Biol. Chem. 2019, 295, 1315–1327. [Google Scholar] [CrossRef]
- Xu, H.; Li, T.; Rohou, A.; Arthur, C.P.; Tzakoniati, F.; Wong, E.; Estevez, A.; Kugel, C.; Franke, Y.; Chen, J.; et al. Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 2019, 176, 702.e14–715.e14. [Google Scholar] [CrossRef]
- Kalina, R.S.; Koshelev, S.G.; Zelepuga, E.; Kim, N.Y.; Kozlov, S.A.; Kozlovskaya, E.; Monastyrnaya, M.; Gladkikh, I. APETx-Like Peptides from the Sea Anemone Heteractis crispa, Diverse in Their Effect on ASIC1a and ASIC3 Ion Channels. Toxins 2020, 12, 266. [Google Scholar] [CrossRef]
- Dutertre, S.; Nicke, A.; Tyndall, J.D.A.; Lewis, R.J. Determination ofα-conotoxin binding modes on neuronal nicotinic acetylcholine receptors. J. Mol. Recognit. 2004, 17, 339–347. [Google Scholar] [CrossRef]
- Dutertre, S.; Nicke, A.; Lewis, R.J. β2 Subunit Contribution to 4/7 α-Conotoxin Binding to the Nicotinic Acetylcholine Receptor. J. Biol. Chem. 2005, 280, 30460–30468. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xu, M.; Zhu, X.; Wu, Y.; Liu, X.; Zhangsun, D.; Hu, Y.; Xiang, S.-H.; Kasheverov, I.E.; Tsetlin, V.I.; et al. From crystal structure of α-conotoxin GIC in complex with Ac-AChBP to molecular determinants of its high selectivity for α3β2 nAChR. Sci. Rep. 2016, 6, 22349. [Google Scholar] [CrossRef] [PubMed]
- Moyer, B.D.; Murray, J.K.; Ligutti, J.; Andrews, K.; Favreau, P.; Jordan, J.B.; Lee, J.H.; Liu, D.; Long, J.; Sham, K.; et al. Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS ONE 2018, 13, e0196791. [Google Scholar] [CrossRef]
- Anand, P.; Grigoryan, A.; Bhuiyan, M.H.; Ueberheide, B.; Russell, V.; Quinoñez, J.; Moy, P.; Chait, B.T.; Poget, S.F.; Holford, M. Sample Limited Characterization of a Novel Disulfide-Rich Venom Peptide Toxin from Terebrid Marine Snail Terebra variegata. PLoS ONE 2014, 9, e94122. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous Fluid Approach to Solvation Thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102, 3531–3541. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous Fluid Approach to Solvation Thermodynamics. 2. Applications to Simple Fluids. J. Phys. Chem. B 1998, 102, 3542–3550. [Google Scholar] [CrossRef]
- Abel, R.; Salam, N.K.; Shelley, J.; Farid, R.; Friesner, R.A.; Sherman, W. Contribution of Explicit Solvent Effects to the Binding Affinity of Small-Molecule Inhibitors in Blood Coagulation Factor Serine Proteases. ChemMedChem 2011, 6, 1049–1066. [Google Scholar] [CrossRef]
- Nguyen, C.N.; Young, T.K.; Gilson, M.K. Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. J. Chem. Phys. 2012, 137, 44101. [Google Scholar] [CrossRef]
- Beglov, D.; Roux, B. An Integral Equation to Describe the Solvation of Polar Molecules in Liquid Water. J. Phys. Chem. B 1997, 101, 7821–7826. [Google Scholar] [CrossRef]
- Kovalenko, A.; Hirata, F. Three-dimensional density profiles of water in contact with a solute of arbitrary shape: A RISM approach. Chem. Phys. Lett. 1998, 290, 237–244. [Google Scholar] [CrossRef]
- Sindhikara, D.; Hirata, F. Analysis of Biomolecular Solvation Sites by 3D-RISM Theory. J. Phys. Chem. B 2013, 117, 6718–6723. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Abel, R.; Kim, B.; Berne, B.J.; Friesner, R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Cappel, D.; Sherman, W.; Beuming, T. Calculating Water Thermodynamics in the Binding Site of Proteins—Applications of WaterMap to Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 2586–2598. [Google Scholar] [CrossRef]
- Beuming, T.; Farid, R.; Sherman, W. High-energy water sites determine peptide binding affinity and specificity of PDZ domains. Protein Sci. 2009, 18, 1609–1619. [Google Scholar] [CrossRef]
- Nittinger, E.; Gibbons, P.; Eigenbrot, C.; Davies, D.R.; Maurer, B.; Yu, C.L.; Kiefer, J.R.; Kuglstatter, A.; Murray, J.; Ortwine, D.F.; et al. Water molecules in protein–ligand interfaces. Evaluation of software tools and SAR comparison. J. Comput. Mol. Des. 2019, 33, 307–330. [Google Scholar] [CrossRef]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R.A. Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [Google Scholar] [CrossRef]
- Higgs, C.; Beuming, T.; Sherman, W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [Google Scholar] [CrossRef]
- Clark, A.J.; Negron, C.; Hauser, K.; Sun, M.; Wang, L.; Abel, R.; Friesner, R.A. Relative Binding Affinity Prediction of Charge-Changing Sequence Mutations with FEP in Protein–Protein Interfaces. J. Mol. Biol. 2019, 431, 1481–1493. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [PubMed]
- Leger, P.R.; Hu, D.X.; Biannic, B.; Bui, M.; Han, X.; Karbarz, E.; Maung, J.; Okano, A.; Osipov, M.; Shibuya, G.M.; et al. Discovery of Potent, Selective, and Orally Bioavailable Inhibitors of USP7 with In Vivo Antitumor Activity. J. Med. Chem. 2020, 63, 5398–5420. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Chang, G.; Spencer, R.H. Crystallographic analyses of ion channels: Lessons and challenges. J. Biol. Chem. 2000, 275, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lee, A.; Campbell, E.; MacKinnon, R.; Kuriyan, J. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2013, 2, e00594. [Google Scholar] [CrossRef]
- Baconguis, I.; Gouaux, E. Structural plasticity and dynamic selectivity of acid-sensing ion channel–spider toxin complexes. Nat. Cell Biol. 2012, 489, 400–405. [Google Scholar] [CrossRef]
- Lau, H.Y.; Hunter, M.J.; Stewart, A.G.; Perozo, E.; Vandenberg, J.I. Never at rest: Insights into the conformational dynamics of ion channels from cryo-electron microscopy. J. Physiol. 2018, 596, 1107–1119. [Google Scholar] [CrossRef]
- Gharpure, A.; Noviello, C.M.; Hibbs, R.E. Progress in nicotinic receptor structural biology. Neuropharmacology 2020, 171, 108086. [Google Scholar] [CrossRef]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nat. Cell Biol. 2016, 534, 347–351. [Google Scholar] [CrossRef]
- Rahman, M.; Teng, J.; Worrell, B.T.; Noviello, C.M.; Lee, M.; Karlin, A.; Stowell, M.H.; Hibbs, R.E. Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins. Neuron 2020, 106, 952–962.e5. [Google Scholar] [CrossRef]
- Cristofori-Armstrong, B.; Saez, N.J.; Chassagnon, I.; King, G.F.; Rash, L.D. The modulation of acid-sensing ion channel 1 by PcTx1 is pH-, subtype- and species-dependent: Importance of interactions at the channel subunit interface and potential for engineering selective analogues. Biochem. Pharmacol. 2019, 163, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; MacKinnon, R. Functional Analysis of Kv1.2 and Paddle Chimera Kv Channels in Planar Lipid Bilayers. J. Mol. Biol. 2008, 382, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A Bivalent Tarantula Toxin Activates the Capsaicin Receptor, TRPV1, by Targeting the Outer Pore Domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef]
- Servent, D.; Winckler-Dietrich, V.; Hu, H.-Y.; Kessler, P.; Drevet, P.; Bertrand, D.; Ménez, A. Only Snake Curaremimetic Toxins with a Fifth Disulfide Bond Have High Affinity for the Neuronal α7 Nicotinic Receptor. J. Biol. Chem. 1997, 272, 24279–24286. [Google Scholar] [CrossRef]
- Henriques, S.T.; Deplazes, E.; Lawrence, N.; Cheneval, O.; Chaousis, S.; Inserra, M.; Thongyoo, P.; King, G.F.; Mark, A.E.; Vetter, I.; et al. Interaction of Tarantula Venom Peptide ProTx-II with Lipid Membranes Is a Prerequisite for Its Inhibition of Human Voltage-gated Sodium Channel NaV1.7. J. Biol. Chem. 2016, 291, 17049–17065. [Google Scholar] [CrossRef]
- Sarkar, D.; Singh, Y.; Kalia, J. Protein–Lipid Interfaces Can Drive the Functions of Membrane-Embedded Protein–Protein Complexes. ACS Chem. Biol. 2018, 13, 2689–2698. [Google Scholar] [CrossRef]
- Rosenthal, J.A.; Levandoski, M.M.; Chang, B.; Potts, J.F.; Shi, Q.-L.; Hawrot, E. The Functional Role of Positively Charged Amino Acid Side Chains in α-Bungarotoxin Revealed by Site-Directed Mutagenesis of a His-Tagged Recombinant α-Bungarotoxin. Biochemistry 1999, 38, 7847–7855. [Google Scholar] [CrossRef]
- Hidalgo, P.; MacKinnon, R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science 1995, 268, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Wonnacott, S.; Barik, J. Nicotinic ACh Receptors. Tocris Rev. 2007, 28, 1–20. [Google Scholar]
- Kohlmann, A.; Zhu, X.; Dalgarno, D. Application of MM-GB/SA and WaterMap to SRC Kinase Inhibitor Potency Prediction. ACS Med. Chem. Lett. 2012, 3, 94–99. [Google Scholar] [CrossRef]
- Flinspach, M.; Xu, Q.; Piekarz, A.D.; Fellows, R.; Hagan, R.; Gibbs, A.C.; Liu, Y.; Neff, R.A.; Freedman, J.; Eckert, W.A.; et al. Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci. Rep. 2017, 7, 39662. [Google Scholar] [CrossRef]
- Jensen, M.Ø.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of Voltage Gating in Potassium Channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef]
- Dawson, R.J.P.; Benz, J.; Stohler, P.; Tetaz, T.; Joseph, C.; Huber, S.; Schmid, G.; Hügin, D.; Pflimlin, P.; Trube, G.; et al. Structure of the Acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat. Commun. 2012, 3, 936. [Google Scholar] [CrossRef]
- Xu, Q.; Tae, H.S.; Wang, Z.; Jiang, T.; Adams, D.J.; Yu, R. Rational Design of alpha-Conotoxin RegIIA Analogues Specifically Inhibiting the Human alpha3beta2 Nicotinic Acetylcholine Receptor through Computational Scanning. ACS Chem. Neurosci. 2020, 2804–2811. [Google Scholar] [CrossRef]
- Long, T.; McDougal, O.M.; Andersen, T. GAMPMS: Genetic algorithm managed peptide mutant screening. J. Comput. Chem. 2015, 36, 1304–1310. [Google Scholar] [CrossRef]
- Rashid, M.H.; Heinzelmann, G.; Huq, R.; Tajhya, R.B.; Chang, S.C.; Chhabra, S.; Pennington, M.W.; Beeton, C.; Norton, R.S.; Kuyucak, S. A Potent and Selective Peptide Blocker of the Kv1.3 Channel: Prediction from Free-Energy Simulations and Experimental Confirmation. PLoS ONE 2013, 8, e78712. [Google Scholar] [CrossRef]
- Ahuja, S.; Mukund, S.; Deng, L.; Khakh, K.; Chang, E.; Ho, H.; Shriver, S.; Young, C.; Lin, S.; Johnson, J.P.; et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350, aac5464. [Google Scholar] [CrossRef]
- Wulff, H.; Christophersen, P.; Colussi, P.; Chandy, K.G.; Yarov-Yarovoy, V. Antibodies and venom peptides: New modalities for ion channels. Nat. Rev. Drug Discov. 2019, 18, 339–357. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion Channel | Peptide Toxin | Structure Information | Potency of Peptide at Channel |
|---|---|---|---|
| Gallus gallus Acid-sensing ion channel 1 (ASIC1a) | Psalmopoeus cambridgei Psalmotoxin-1 (PcTx1) | Xtal, 3.4Å, (PDB: 4FZ1) [35] | IC50 = 3 nM (Human ASIC1a) [41] |
| Rattus norvegicus Kv1.2-2.1 paddle chimera channel (Kv) | Leiurus hebraeus Charybdotoxin (CTX) | Xtal, 2.5Å, (PDB: 4JTA) [34] | KD = 19 nM [34,42] |
| Homo sapiens/Arcobacter butzleri VSD2-NavAb channel chimera (Nav) | Thrixopelma pruriens β/ω theraphotoxin-Tp2a (ProTx2) | Activated: Cryo-EM, 3.6Å (PDB: 6N4Q) Deactivated: Cryo-EM, 4.2Å (PDB: 6N4R) [8] | IC50 = 0.26 nM [8] |
| Rattus norvegicus Transient receptor potential cation channel subfamily V member 1 (TRPV1) | Haplopelma schmidti Double-knot toxin (DkTx) | Cryo-EM, 3.0Å, (PDB: 5IRX) [39] | EC50 = 0.23 µM [43] |
| Tetronarce californica Nicotinic receptor (nAChR) | Bungarus multicinctus α-bungarotoxin (BTX) | Cryo-EM, 2.7Å, (PDB: 6UWZ) [40] | KD = 0.4 nM [44] |
| Selected Peptide Toxin Residue | Overlap with Unstable Water Site? | Mutation | Fold Loss in Potency of Mutant or Presumed Functional Role | |
|---|---|---|---|---|
| PcTx1 [35] | W7 | Yes | W7A | >318X [41] 1 |
| R27 | Yes | R27A | 11X [41] 1 | |
| W24 | No | W24A | >318X [41] 1 | |
| R26 | No | R26A | 52X [41] 1 | |
| CTX [34] | K27 | Yes | K27M | 33X [34] |
| ProTx2 [8] | W7 | Yes | W7Y | 112X [45] 2 |
| W24 | Yes | W24Y | 181X [45] 2 | |
| L29 | Yes | N/A | Anchors ProTx2 in membrane [8] | |
| W5 | No | W5Y | 292X [45] 2 | |
| R22 | No | R22E | >1000X [8] | |
| K26 | No | K26D | >100X [8] | |
| W30 | No | W30Y | 6X [45] 2 | |
| W30L | 22X [45] 2 | |||
| DkTx [39] | W11 | Yes | W11A | 80X [46] |
| W11L | 43X [46] | |||
| W11Y | 9X [46] | |||
| F27 | Yes | F27A | 7X [46] | |
| W53 | Yes | W53A | >100X [46] | |
| W53L | >100X [46] | |||
| W53Y | 8X [46] | |||
| F67 | Yes | F67A | 13X [46] | |
| BTX [40] | F32 | Yes | N/A | Cation-π stack with R36 [40] |
| R36 | Yes | R36A | 90X [47] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, B.; Sindhikara, D.; Borrelli, K.; Leffler, A.E. Water Thermodynamics of Peptide Toxin Binding Sites on Ion Channels. Toxins 2020, 12, 652. https://doi.org/10.3390/toxins12100652
Shah B, Sindhikara D, Borrelli K, Leffler AE. Water Thermodynamics of Peptide Toxin Binding Sites on Ion Channels. Toxins. 2020; 12(10):652. https://doi.org/10.3390/toxins12100652
Chicago/Turabian StyleShah, Binita, Dan Sindhikara, Ken Borrelli, and Abba E. Leffler. 2020. "Water Thermodynamics of Peptide Toxin Binding Sites on Ion Channels" Toxins 12, no. 10: 652. https://doi.org/10.3390/toxins12100652
APA StyleShah, B., Sindhikara, D., Borrelli, K., & Leffler, A. E. (2020). Water Thermodynamics of Peptide Toxin Binding Sites on Ion Channels. Toxins, 12(10), 652. https://doi.org/10.3390/toxins12100652