Modifying Phosphate Toxicity in Chronic Kidney Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Formation of Calciprotein Particles
3. FGF23 as Mediator of Phosphate Toxicity
4. Mitigating Effects of Magnesium on Phosphate Toxicity
5. The Role of Calcium
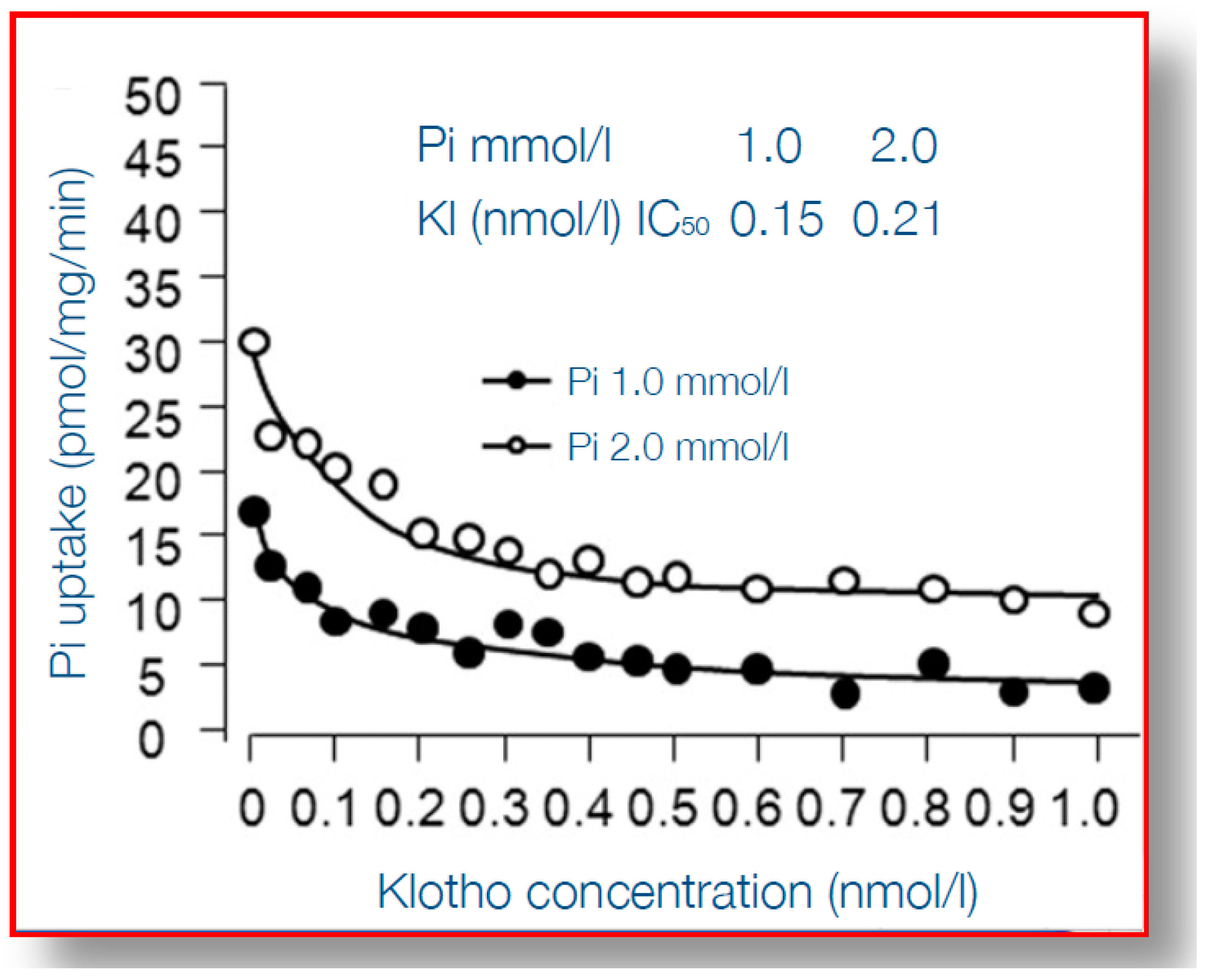
6. α-Klotho Mitigates Phosphate Toxicity
7. Matrix Gla Protein and Vitamin K Status
8. Additional Factors that May Modify Phosphate-Toxicity
9. Clinical Implications for Alleviating Phosphate-Induced Complications
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D.; ERA–EDTA Working Group on Chronic Kidney Disease–Mineral and Bone Disorders and the European Renal Nutrition Working Group. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2016. [Google Scholar] [CrossRef]
- Gattineni, J.; Baum, M. Genetic disorders of phosphate regulation. Pediatr. Nephrol. 2012, 27, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef]
- Chiu, Y.W.; Teitelbaum, I.; Misra, M.; de Leon, E.M.; Adzize, T.; Mehrotra, R. Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1089–1096. [Google Scholar] [CrossRef]
- Vervloet, M.G.; van Ballegooijen, A.J. Prevention and treatment of hyperphosphatemia in chronic kidney disease. Kidney Int. 2018, 93, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Danese, M.D.; Belozeroff, V.; Smirnakis, K.; Rothman, K.J. Consistent control of mineral and bone disorder in incident hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Karavetian, M.; de Vries, N.; Rizk, R.; Elzein, H. Dietary educational interventions for management of hyperphosphatemia in hemodialysis patients: A systematic review and meta-analysis. Nutr. Rev. 2014, 72, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, S.; Castelino, R.L.; Lioufas, N.M.; Peterson, G.M.; Zaidi, S.T. Nonadherence to Medication Therapy in Haemodialysis Patients: A Systematic Review. PLoS ONE 2015, 10, e0144119. [Google Scholar] [CrossRef]
- Karamanidou, C.; Clatworthy, J.; Weinman, J.; Horne, R. A systematic review of the prevalence and determinants of nonadherence to phosphate binding medication in patients with end-stage renal disease. BMC Nephrol. 2008, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Hugtenburg, J.G.; Timmers, L.; Elders, P.J.; Vervloet, M.; van Dijk, L. Definitions, variants, and causes of nonadherence with medication: A challenge for tailored interventions. Patient Prefer Adherence 2013, 7, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Pipich, V.; Jahnen-Dechent, W.; Schwahn, D. Fetuin-A is a mineral carrier protein: Small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys. J. 2010, 99, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Gangneux, C.; Daveau, M.; Hiron, M.; Derambure, C.; Papaconstantinou, J.; Salier, J.P. The inflammation-induced down-regulation of plasma Fetuin-A (alpha2HS-Glycoprotein) in liver results from the loss of interaction between long C/EBP isoforms at two neighbouring binding sites. Nucleic Acids Res. 2003, 31, 5957–5970. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, E.R.; Hanssen, E.; McMahon, L.P.; Holt, S.G. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PLoS ONE 2013, 8, e60904. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-alpha. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Rajkumar, C.; McMahon, L.P.; Holt, S.G. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2012, 27, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Bohm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Hamano, T.; Matsui, I.; Mikami, S.; Tomida, K.; Fujii, N.; Imai, E.; Rakugi, H.; Isaka, Y. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J. Am. Soc. Nephrol. 2010, 21, 1998–2007. [Google Scholar] [CrossRef]
- Holt, S.G.; Smith, E.R. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2016, 31, 1583–1587. [Google Scholar] [CrossRef]
- Pasch, A.; Farese, S.; Graber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Keyzer, C.A.; de Borst, M.H.; van den Berg, E.; Jahnen-Dechent, W.; Arampatzis, S.; Farese, S.; Bergmann, I.P.; Floege, J.; Navis, G.; Bakker, S.J.; et al. Calcification Propensity and Survival among Renal Transplant Recipients. J. Am. Soc. Nephrol. 2016, 27, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Block, G.A.; Bachtler, M.; Smith, E.R.; Jahnen-Dechent, W.; Arampatzis, S.; Chertow, G.M.; Parfrey, P.; Ma, X.; Floege, J. Blood Calcification Propensity, Cardiovascular Events, and Survival in Patients Receiving Hemodialysis in the EVOLVE Trial. Clin. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Bodenham, E.; McMahon, L.P.; Farese, S.; Rajkumar, C.; Holt, S.G.; Pasch, A. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J. Am. Soc. Nephrol. 2014, 25, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A. Novel assessments of systemic calcification propensity. Curr. Opin. Nephrol. Hypertens. 2016, 25, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Calciprotein particle (CPP): A true culprit of phosphorus woes? Nefrologia 2014, 34, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortiz, M.E.; Lopez, I.; Munoz-Castaneda, J.R.; Martinez-Moreno, J.M.; Ramirez, A.P.; Pineda, C.; Canalejo, A.; Jaeger, P.; Aguilera-Tejero, E.; Rodriguez, M.; et al. Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef]
- Carrillo-Lopez, N.; Panizo, S.; Alonso-Montes, C.; Roman-Garcia, P.; Rodriguez, I.; Martinez-Salgado, C.; Dusso, A.S.; Naves, M.; Cannata-Andia, J.B. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. 2016, 90, 77–89. [Google Scholar] [CrossRef]
- Vervloet, M.G.; van Ittersum, F.J.; Buttler, R.M.; Heijboer, A.C.; Blankenstein, M.A.; ter Wee, P.M. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin. J. Am. Soc. Nephrol. 2011, 6, 383–389. [Google Scholar] [CrossRef]
- Wolf, M. Forging forward with 10 burning questions on FGF23 in kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Verkaik, M.; Oranje, M.; Abdurrachim, D.; Goebel, M.; Gam, Z.; Prompers, J.J.; Helmes, M.; Ter Wee, P.M.; van der Velden, J.; Kuster, D.W.; et al. High Fibroblast Growth Factor 23 concentrations in experimental renal failure impair calcium handling in cardiomyocytes. Physiol. Rep. 2018, 6, e13591. [Google Scholar] [CrossRef] [PubMed]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef] [PubMed]
- Faul, C. FGF23 effects on the heart-levels, time, source, and context matter. Kidney Int. 2018, 94, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Hogler, W.; Linglart, A.; Padidela, R.; Van’t Hoff, W.; Mao, M.; Chen, C.Y.; et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef]
- Insogna, K.L.; Briot, K.; Imel, E.A.; Kamenicky, P.; Ruppe, M.D.; Portale, A.A.; Weber, T.; Pitukcheewanont, P.; Cheong, H.I.; Jan de Beur, S.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J. Bone Min. Res. 2018, 33, 1383–1393. [Google Scholar] [CrossRef]
- Moe, S.M.; Chertow, G.M.; Parfrey, P.S.; Kubo, Y.; Block, G.A.; Correa-Rotter, R.; Drueke, T.B.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis: The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Circulation 2015, 132, 27–39. [Google Scholar] [CrossRef]
- Liu, Y.L.; Huang, C.C.; Chang, C.C.; Chou, C.Y.; Lin, S.Y.; Wang, I.K.; Hsieh, D.J.; Jong, G.P.; Huang, C.Y.; Wang, C.M. Hyperphosphate-Induced Myocardial Hypertrophy through the GATA-4/NFAT-3 Signaling Pathway Is Attenuated by ERK Inhibitor Treatment. Cardiorenal Med. 2015, 5, 79–88. [Google Scholar] [CrossRef]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef]
- Scialla, J.J.; Xie, H.; Rahman, M.; Anderson, A.H.; Isakova, T.; Ojo, A.; Zhang, X.; Nessel, L.; Hamano, T.; Grunwald, J.E.; et al. Fibroblast Growth Factor-23 and Cardiovascular Events in CKD. J. Am. Soc. Nephrol. 2014, 25, 349–360. [Google Scholar] [CrossRef]
- Verkaik, M.; Juni, R.P.; van Loon, E.P.M.; van Poelgeest, E.; Kwekkeboom, R.F.J.; Gam, Z.; Richards, W.G.; Ter Wee, P.M.; Hoenderop, J.G.J.; Eringa, E.C.; et al. FGF23 impairs peripheral microvascular function in renal failure. Am. J. Physiol. Heart Circ. Physiol. 2018. [Google Scholar] [CrossRef]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Metzinger-Le Meuth, V.; Henaut, L.; Djelouat, M.S.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: Evidence for the involvement of miR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef]
- Gross, P.; Six, I.; Kamel, S.; Massy, Z.A. Vascular toxicity of phosphate in chronic kidney disease: Beyond vascular calcification. Circ. J. 2014, 78, 2339–2346. [Google Scholar] [CrossRef]
- Di Marco, G.S.; Reuter, S.; Kentrup, D.; Grabner, A.; Amaral, A.P.; Fobker, M.; Stypmann, J.; Pavenstadt, H.; Wolf, M.; Faul, C.; et al. Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2014, 29, 2028–2035. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Fujii, N.; Shoji, T.; Hayashi, T.; Rakugi, H.; Isaka, Y. Hypomagnesemia is a significant predictor of cardiovascular and non-cardiovascular mortality in patients undergoing hemodialysis. Kidney Int. 2014, 85, 174–181. [Google Scholar] [CrossRef]
- De Roij van Zuijdewijn, C.L.; Grooteman, M.P.; Bots, M.L.; Blankestijn, P.J.; Steppan, S.; Buchel, J.; Groenwold, R.H.; Brandenburg, V.; van den Dorpel, M.A.; Ter Wee, P.M.; et al. Serum Magnesium and Sudden Death in European Hemodialysis Patients. PLoS ONE 2015, 10, e0143104. [Google Scholar] [CrossRef]
- Leenders, N.H.J.; Vervloet, M.G. Magnesium: A Magic Bullet for Cardiovascular Disease in Chronic Kidney Disease? Nutrients 2019, 11, 455. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Fujii, N.; Shoji, T.; Hayashi, T.; Rakugi, H.; Iseki, K.; Tsubakihara, Y.; Isaka, Y.; Committee of Renal Data Registry of the Japanese Society for Dialysis Therapy. Magnesium modifies the cardiovascular mortality risk associated with hyperphosphatemia in patients undergoing hemodialysis: A cohort study. PLoS ONE 2014, 9, e116273. [Google Scholar] [CrossRef]
- Mizuiri, S.; Nishizawa, Y.; Yamashita, K.; Naito, T.; Ono, K.; Tanji, C.; Usui, K.; Doi, S.; Masaki, T.; Shigemoto, K. Hypomagnesemia is not an independent risk factor for mortality in Japanese maintenance hemodialysis patients. Int. Urol. Nephrol. 2019, 51, 1043–1052. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Obi, Y.; Monden, C.; Oka, T.; Yamaguchi, S.; Matsui, I.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J. Am. Soc. Nephrol. 2019, 30, 1073–1085. [Google Scholar] [CrossRef]
- Joris, P.J.; Plat, J.; Bakker, S.J.; Mensink, R.P. Long-term magnesium supplementation improves arterial stiffness in overweight and obese adults: Results of a randomized, double-blind, placebo-controlled intervention trial. Am. J. Clin. Nutr. 2016, 103, 1260–1266. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular calcification: In vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. 2003, 14, S300–S304. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Louvet, L.; Buchel, J.; Steppan, S.; Passlick-Deetjen, J.; Massy, Z.A. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2013, 28, 869–878. [Google Scholar] [CrossRef]
- Montes de Oca, A.; Guerrero, F.; Martinez-Moreno, J.M.; Madueno, J.A.; Herencia, C.; Peralta, A.; Almaden, Y.; Lopez, I.; Aguilera-Tejero, E.; Gundlach, K.; et al. Magnesium inhibits Wnt/beta-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS ONE 2014, 9, e89525. [Google Scholar] [CrossRef]
- Tomazic, B.; Tomson, M.; Nancollas, G.H. Growth of calcium phosphates on hydroxyapatite crystals: The effect of magnesium. Arch. Oral Biol. 1975, 20, 803–808. [Google Scholar] [CrossRef]
- Ter Braake, A.D.; Tinnemans, P.T.; Shanahan, C.M.; Hoenderop, J.G.J.; de Baaij, J.H.F. Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci. Rep. 2018, 8, 2069. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Silver, B.; Pasch, A.; Bouchelouche, P.; Pedersen, L.; Rasmussen, L.M.; Brandi, L. Oral Magnesium Supplementation in Chronic Kidney Disease Stages 3 and 4: Efficacy, Safety, and Effect on Serum Calcification Propensity-A Prospective Randomized Double-Blinded Placebo-Controlled Clinical Trial. Kidney Int. Rep. 2017, 2, 380–389. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Pasch, A.; Brandi, L. The Effect of Increasing Dialysate Magnesium on Serum Calcification Propensity in Subjects with End Stage Kidney Disease: A Randomized, Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2018, 13, 1373–1380. [Google Scholar] [CrossRef]
- Diaz-Tocados, J.M.; Peralta-Ramirez, A.; Rodriguez-Ortiz, M.E.; Raya, A.I.; Lopez, I.; Pineda, C.; Herencia, C.; Montes de Oca, A.; Vergara, N.; Steppan, S.; et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int. 2017, 92, 1084–1099. [Google Scholar] [CrossRef]
- Massy, Z.A.; Nistor, I.; Apetrii, M.; Brandenburg, V.M.; Bover, J.; Evenepoel, P.; Goldsmith, D.; Mazzaferro, S.; Urena-Torres, P.; Vervloet, M.G.; et al. Magnesium-based interventions for normal kidney function and chronic kidney disease. Magnes. Res. 2016, 29, 126–140. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Ewence, A.E.; Bootman, M.; Roderick, H.L.; Skepper, J.N.; McCarthy, G.; Epple, M.; Neumann, M.; Shanahan, C.M.; Proudfoot, D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: A potential mechanism in atherosclerotic plaque destabilization. Circ. Res. 2008, 103, e28–e34. [Google Scholar] [CrossRef]
- Han, N.; Hong, S.H.; Kim, Y.S.; Kim, D.K.; Kim, I.W.; Ji, E.; Oh, J.M. Effect of additive calcium administration on FGF23 levels in patients with mild chronic kidney disease treated with calcitriol: A randomized, open-labeled clinical trial. Clin. Risk Manag. 2017, 13, 999–1007. [Google Scholar] [CrossRef]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef]
- Block, G.A.; Raggi, P.; Bellasi, A.; Kooienga, L.; Spiegel, D.M. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007, 71, 438–441. [Google Scholar] [CrossRef]
- Olauson, H.; Mencke, R.; Hillebrands, J.L.; Larsson, T.E. Tissue expression and source of circulating alphaKlotho. Bone 2017, 100, 19–35. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Farrow, E.G.; Davis, S.I.; Summers, L.J.; White, K.E. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J. Am. Soc. Nephrol. 2009, 20, 955–960. [Google Scholar] [CrossRef]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Hum, J.M.; O’Bryan, L.M.; Tatiparthi, A.K.; Cass, T.A.; Clinkenbeard, E.L.; Cramer, M.S.; Bhaskaran, M.; Johnson, R.L.; Wilson, J.M.; Smith, R.C.; et al. Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Renal and extrarenal actions of Klotho. Semin. Nephrol. 2013, 33, 118–129. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Addo, T.; Cho, H.J.; Barker, S.L.; Ravikumar, P.; Gillings, N.; Bian, A.; Sidhu, S.S.; et al. Renal Production, Uptake, and Handling of Circulating alphaKlotho. J. Am. Soc. Nephrol. 2016, 27, 79–90. [Google Scholar] [CrossRef]
- Andrukhova, O.; Bayer, J.; Schuler, C.; Zeitz, U.; Murali, S.K.; Ada, S.; Alvarez-Pez, J.M.; Smorodchenko, A.; Erben, R.G. Klotho Lacks an FGF23-Independent Role in Mineral Homeostasis. J. Bone Min. Res. 2017, 32, 2049–2061. [Google Scholar] [CrossRef]
- Sakan, H.; Nakatani, K.; Asai, O.; Imura, A.; Tanaka, T.; Yoshimoto, S.; Iwamoto, N.; Kurumatani, N.; Iwano, M.; Nabeshima, Y.; et al. Reduced renal alpha-Klotho expression in CKD patients and its effect on renal phosphate handling and vitamin D metabolism. PLoS ONE 2014, 9, e86301. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.L.; Pastor, J.; Carranza, D.; Quinones, H.; Griffith, C.; Goetz, R.; Mohammadi, M.; Ye, J.; Zhang, J.; Hu, M.C.; et al. The demonstration of alphaKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2015, 30, 223–233. [Google Scholar] [CrossRef]
- Young, G.H.; Wu, V.C. KLOTHO methylation is linked to uremic toxins and chronic kidney disease. Kidney Int. 2012, 81, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; Adema, A.Y.; Larsson, T.E.; Massy, Z.A. The role of klotho on vascular calcification and endothelial function in chronic kidney disease. Semin. Nephrol. 2014, 34, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Nakatani, T.; Lanske, B.; Razzaque, M.S. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-dihydroxyvitamin d levels. Circ. Cardiovasc. Genet. 2009, 2, 583–590. [Google Scholar] [CrossRef]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010, 24, 3562–3571. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Kinoshita, S.; Ozono, K.; Michigami, T. Inorganic Phosphate Activates the AKT/mTORC1 Pathway and Shortens the Life Span of an alphaKlotho-Deficient Model. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef]
- Mohammad, J.; Scanni, R.; Bestmann, L.; Hulter, H.N.; Krapf, R. A Controlled Increase in Dietary Phosphate Elevates BP in Healthy Human Subjects. J. Am. Soc. Nephrol. 2018, 29, 2089–2098. [Google Scholar] [CrossRef]
- Kusaba, T.; Okigaki, M.; Matui, A.; Murakami, M.; Ishikawa, K.; Kimura, T.; Sonomura, K.; Adachi, Y.; Shibuya, M.; Shirayama, T.; et al. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc. Natl. Acad. Sci. USA 2010, 107, 19308–19313. [Google Scholar] [CrossRef]
- Rattazzi, M.; Bertacco, E.; Del Vecchio, A.; Puato, M.; Faggin, E.; Pauletto, P. Aortic valve calcification in chronic kidney disease. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2013, 28, 2968–2976. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yao, Q.; Ao, L.; Cleveland, J.C., Jr.; Dong, N.; Fullerton, D.A.; Meng, X. Klotho suppresses high phosphate-induced osteogenic responses in human aortic valve interstitial cells through inhibition of Sox9. J. Mol. Med. 2017, 95, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Cha, S.K.; An, S.W.; Kuro, O.M.; Birnbaumer, L.; Huang, C.L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yoon, J.; An, S.W.; Kuro-o, M.; Huang, C.L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J. Am. Soc. Nephrol. 2015, 26, 1150–1160. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Cranenburg, E.C.; Vermeer, C. Matrix Gla-protein: The calcification inhibitor in need of vitamin K. Thromb. Haemost. 2008, 100, 593–603. [Google Scholar] [PubMed]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2016, 31, 31–39. [Google Scholar] [CrossRef]
- Wasilewski, G.B.; Vervloet, M.G.; Schurgers, L.J. The Bone—Vasculature Axis: Calcium Supplementation and the Role of Vitamin K. Front. Cardiovasc. Med. 2019, 6. [Google Scholar] [CrossRef]
- Cranenburg, E.C.; Vermeer, C.; Koos, R.; Boumans, M.L.; Hackeng, T.M.; Bouwman, F.G.; Kwaijtaal, M.; Brandenburg, V.M.; Ketteler, M.; Schurgers, L.J. The circulating inactive form of matrix Gla Protein (ucMGP) as a biomarker for cardiovascular calcification. J. Vasc. Res. 2008, 45, 427–436. [Google Scholar] [CrossRef]
- Han, K.H.; O’Neill, W.C. Increased Peripheral Arterial Calcification in Patients Receiving Warfarin. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Bloch, D.B.; Nazarian, R.M.; Vermeer, C.; Booth, S.L.; Xu, D.; Thadhani, R.I.; Malhotra, R. Vitamin K-Dependent Carboxylation of Matrix Gla Protein Influences the Risk of Calciphylaxis. J. Am. Soc. Nephrol. 2017, 28, 1717–1722. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Renard, C.; Magdeleyns, E.J.; Vermeer, C.; Choukroun, G.; Massy, Z.A. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin. J. Am. Soc. Nephrol. 2010, 5, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J. Role of vitamin K and Gla proteins in the pathophysiology of osteoporosis and vascular calcification. Curr. Opin. Clin. Nutr. Metab. Care 2000, 3, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Tantisattamo, E.; Han, K.H.; O’Neill, W.C. Increased vascular calcification in patients receiving warfarin. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 237–242. [Google Scholar] [CrossRef] [PubMed]
- van Ballegooijen, A.J.; Beulens, J.W. The Role of Vitamin K Status in Cardiovascular Health: Evidence from Observational and Clinical Studies. Curr. Nutr. Rep. 2017, 6, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Krueger, T.; Westenfeld, R.; Ketteler, M.; Schurgers, L.J.; Floege, J. Vitamin K deficiency in CKD patients: A modifiable risk factor for vascular calcification? Kidney Int. 2009, 76, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Krueger, T.; Schlieper, G.; Schurgers, L.; Cornelis, T.; Cozzolino, M.; Jacobi, J.; Jadoul, M.; Ketteler, M.; Rump, L.C.; Stenvinkel, P.; et al. Vitamin K1 to slow vascular calcification in haemodialysis patients (VitaVasK trial): A rationale and study protocol. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2014, 29, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Silaghi, C.N.; Fodor, D.; Gheorghe, S.R.; Craciun, A.M. Serum total matrix Gla protein: Reference interval in healthy adults and variations in patients with vascular and osteoarticular diseases. Clin. Chim. Acta 2019, 490, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Houben, E.; Neradova, A.; Schurgers, L.J.; Vervloet, M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G. Ital. Nefrol. 2016, 33, gin/33.6.5. [Google Scholar] [PubMed]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Sauer, G.R.; Wu, L.N.; Iijima, M.; Wuthier, R.E. The influence of trace elements on calcium phosphate formation by matrix vesicles. J. Inorg. Biochem. 1997, 65, 57–65. [Google Scholar] [CrossRef]
- Shin, M.Y.; Kwun, I.S. Phosphate-induced rat vascular smooth muscle cell calcification and the implication of zinc deficiency in a7r5 cell viability. Prev. Nutr. Food Sci. 2013, 18, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Tuffaha, R.; Luong, T.T.D.; Zickler, D.; Masyout, J.; Feger, M.; Verheyen, N.; Blaschke, F.; Kuro, O.M.; Tomaschitz, A.; et al. Zinc Inhibits Phosphate-Induced Vascular Calcification through TNFAIP3-Mediated Suppression of NF-kappaB. J. Am. Soc. Nephrol. 2018, 29, 1636–1648. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Eisenberg, R.; Mowrey, W.B.; Wylie-Rosett, J.; Abramowitz, M.K.; Bushinsky, D.A.; Melamed, M.L. Association between dietary zinc intake and abdominal aortic calcification in US adults. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Virkki, L.V.; Biber, J.; Murer, H.; Forster, I.C. Phosphate transporters: A tale of two solute carrier families. Am. J. Physiol. Ren. Physiol. 2007, 293, F643–F654. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Leibrock, C.B.; Alesutan, I.; Voelkl, J.; Pakladok, T.; Michael, D.; Schleicher, E.; Kamyabi-Moghaddam, Z.; Quintanilla-Martinez, L.; Kuro-o, M.; Lang, F. NH4Cl Treatment Prevents Tissue Calcification in Klotho Deficiency. J. Am. Soc. Nephrol. 2015, 26, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Leibrock, C.B.; Voelkl, J.; Kohlhofer, U.; Quintanilla-Martinez, L.; Kuro, O.M.; Lang, F. Bicarbonate-sensitive calcification and lifespan of klotho-deficient mice. Am. J. Physiol. Ren. Physiol. 2016, 310, F102–F108. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.J.; Lopez, I.; Montes de Oca, A.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Metabolic acidosis inhibits soft tissue calcification in uremic rats. Kidney Int. 2008, 73, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Ohtake, T.; Mochida, Y.; Ishioka, K.; Maesato, K.; Moriya, H.; Hidaka, S.; Kobayashi, S. Correlation of coronary artery calcification with pre-hemodialysis bicarbonate levels in patients on hemodialysis. Apher. Dial. Off. Peer-Rev. J. Int. Soc. Apher. Jpn. Soc. Apher. Jpn. Soc. Dial. 2012, 16, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Lamina, C.; Kronenberg, F.; Stenvinkel, P.; Froissart, M.; Forer, L.; Schonherr, S.; Wheeler, D.C.; Eckardt, K.U.; Floege, J. Association of changes in bone mineral parameters with mortality in haemodialysis patients: Insights from the ARO cohort. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, M.; Kazama, J.J.; Wada, A.; Hamano, T.; Masakane, I.; Narita, I. Functional impairment attenuates the association between high serum phosphate and mortality in dialysis patients: A nationwide cohort study. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2019, 34, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Bristow, S.M.; Gamble, G.D.; Pasch, A.; O’Neill, W.C.; Stewart, A.; Horne, A.M.; Reid, I.R. Acute and 3-month effects of calcium carbonate on the calcification propensity of serum and regulators of vascular calcification: Secondary analysis of a randomized controlled trial. Osteoporos. Int. 2016, 27, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.; Pasch, A.; van der Sande, F.; Konings, C.; Bachtler, M.; Dionisi, M.; Meier, M.; Kooman, J.; Canaud, B. High-Flux Hemodialysis and High-Volume Hemodiafiltration Improve Serum Calcification Propensity. PLoS ONE 2016, 11, e0151508. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Sprague, S.M.; Covic, A.C.; Rastogi, A.; Spinowitz, B.; Rakov, V.; Walpen, S.; Floege, J. Effects of sucroferric oxyhydroxide and sevelamer carbonate on chronic kidney disease-mineral bone disorder parameters in dialysis patients. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rodelo-Haad, C.; Rodriguez-Ortiz, M.E.; Martin-Malo, A.; Pendon-Ruiz de Mier, M.V.; Aguera, M.L.; Munoz-Castaneda, J.R.; Soriano, S.; Caravaca, F.; Alvarez-Lara, M.A.; Felsenfeld, A.; et al. Phosphate control in reducing FGF23 levels in hemodialysis patients. PLoS ONE 2018, 13, e0201537. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2015. [Google Scholar] [CrossRef]
- Farrow, E.G.; Yu, X.; Summers, L.J.; Davis, S.I.; Fleet, J.C.; Allen, M.R.; Robling, A.G.; Stayrook, K.R.; Jideonwo, V.; Magers, M.J.; et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Matsui, I.; Oka, T.; Yamaguchi, S.; Kubota, K.; Shimada, K.; Matsumoto, A.; Hashimoto, N.; Isaka, Y. Low magnesium diet aggravates phosphate-induced kidney injury. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2018. [Google Scholar] [CrossRef]
- Leenders, N.H.J.; van Ittersum, F.J.; Hoekstra, T.; Hoenderop, J.G.J.; Vervloet, M.G. Routine hemodialysis induces a decline in plasma magnesium concentration in most patients: A prospective observational cohort study. Sci. Rep. 2018, 8, 10256. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Kragelund, C.; Brandi, L. The effect of magnesium supplementation on vascular calcification in chronic kidney disease-a randomised clinical trial (MAGiCAL-CKD): Essential study design and rationale. BMJ Open 2017, 7, e016795. [Google Scholar] [CrossRef]
- Zhang, Q.; Yin, S.; Liu, L.; Liu, Z.; Cao, W. Rhein reversal of DNA hypermethylation-associated Klotho suppression ameliorates renal fibrosis in mice. Sci. Rep. 2016, 6, 34597. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Xu, Y.; Sun, Z. Induction of anti-aging gene klotho with a small chemical compound that demethylates CpG islands. Oncotarget 2017, 8, 46745–46755. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Mamiya, H.; Shinde, S.N.; Cheikhi, A.; Winter, L.L.; Vo, N.V.; Stolz, D.; Roginskaya, V.; Tang, W.Y.; St Croix, C.; et al. Age-related declines in alpha-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat. Commun. 2018, 9, 4859. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.M.; Kuro-o, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Henriquez-Palop, F.; Martin-Nunez, E.; Perez-Delgado, N.; Muros-de-Fuentes, M.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Effect of Paricalcitol on FGF-23 and Klotho in Kidney Transplant Recipients. Transplantation 2016, 100, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.; Brandenburg, V.M.; Vermeer, C.; Stenger, M.; Muhlenbruch, G.; Mahnken, A.H.; Gladziwa, U.; Ketteler, M.; Schurgers, L.J. Uncarboxylated matrix Gla protein (ucMGP) is associated with coronary artery calcification in haemodialysis patients. Thromb. Haemost. 2009, 101, 359–366. [Google Scholar] [PubMed]
- Shea, M.K.; O’Donnell, C.J.; Hoffmann, U.; Dallal, G.E.; Dawson-Hughes, B.; Ordovas, J.M.; Price, P.A.; Williamson, M.K.; Booth, S.L. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am. J. Clin. Nutr. 2009, 89, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Reinartz, S.; Kaesler, N.; Kruger, T.; Dirrichs, T.; Kramann, R.; Peeters, F.; Floege, J.; Keszei, A.; Marx, N.; et al. Slower Progress of Aortic Valve Calcification with Vitamin K Supplementation: Results from a Prospective Interventional Proof-of-Concept Study. Circulation 2017, 135, 2081–2083. [Google Scholar] [CrossRef]
- Caluwe, R.; Pyfferoen, L.; De Boeck, K.; De Vriese, A.S. The effects of vitamin K supplementation and vitamin K antagonists on progression of vascular calcification: Ongoing randomized controlled trials. Clin. Kidney J. 2016, 9, 273–279. [Google Scholar] [CrossRef]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vervloet, M. Modifying Phosphate Toxicity in Chronic Kidney Disease. Toxins 2019, 11, 522. https://doi.org/10.3390/toxins11090522
Vervloet M. Modifying Phosphate Toxicity in Chronic Kidney Disease. Toxins. 2019; 11(9):522. https://doi.org/10.3390/toxins11090522
Chicago/Turabian StyleVervloet, Marc. 2019. "Modifying Phosphate Toxicity in Chronic Kidney Disease" Toxins 11, no. 9: 522. https://doi.org/10.3390/toxins11090522
APA StyleVervloet, M. (2019). Modifying Phosphate Toxicity in Chronic Kidney Disease. Toxins, 11(9), 522. https://doi.org/10.3390/toxins11090522