Effects of Predator-Prey Interactions on Predator Traits: Differentiation of Diets and Venoms of a Marine Snail


Abstract
:1. Introduction
2. Results
2.1. Dietary Characterizations
2.2. Sequence Datasets, de novo Transcriptome Assembly and Transcript Annotation
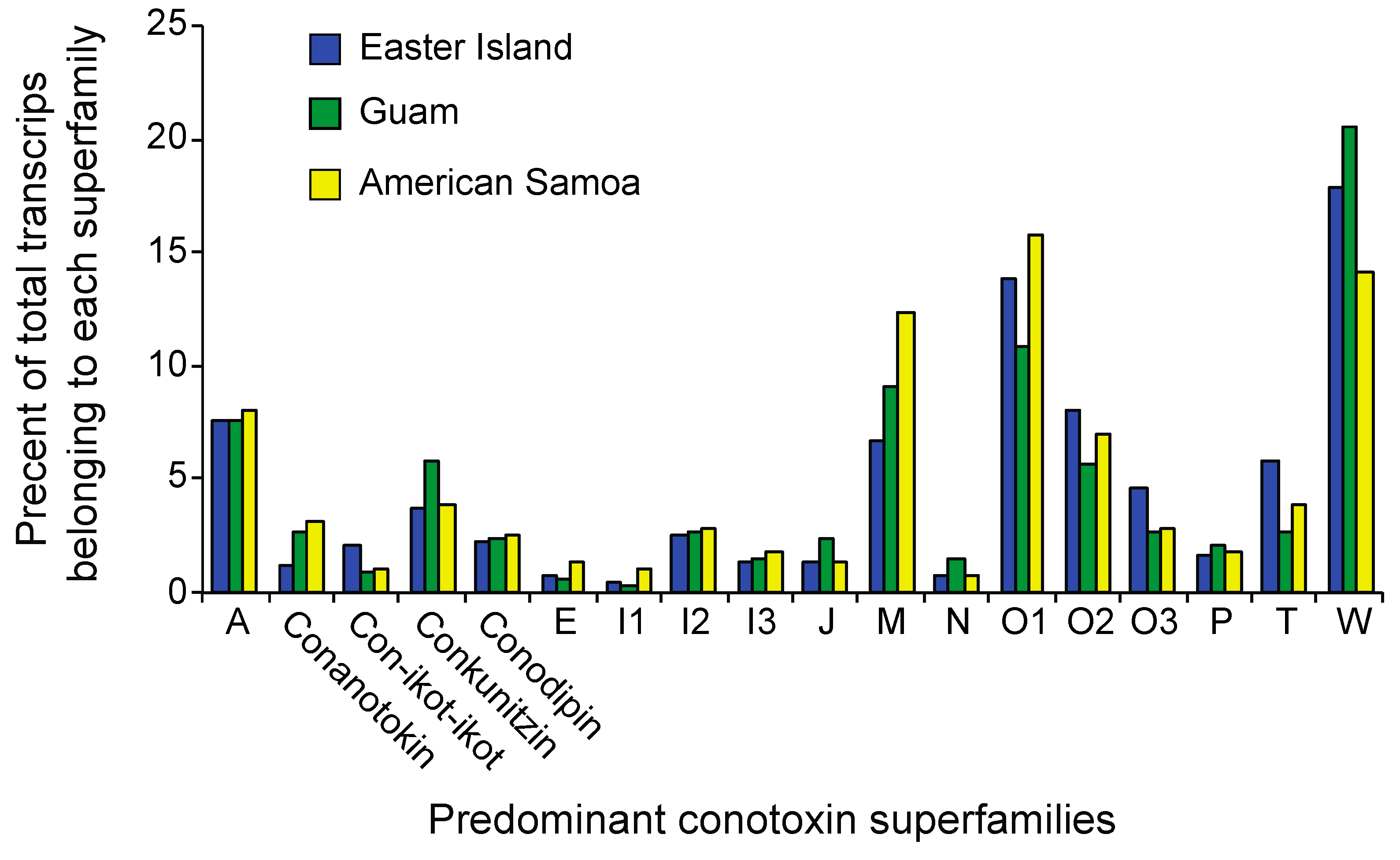
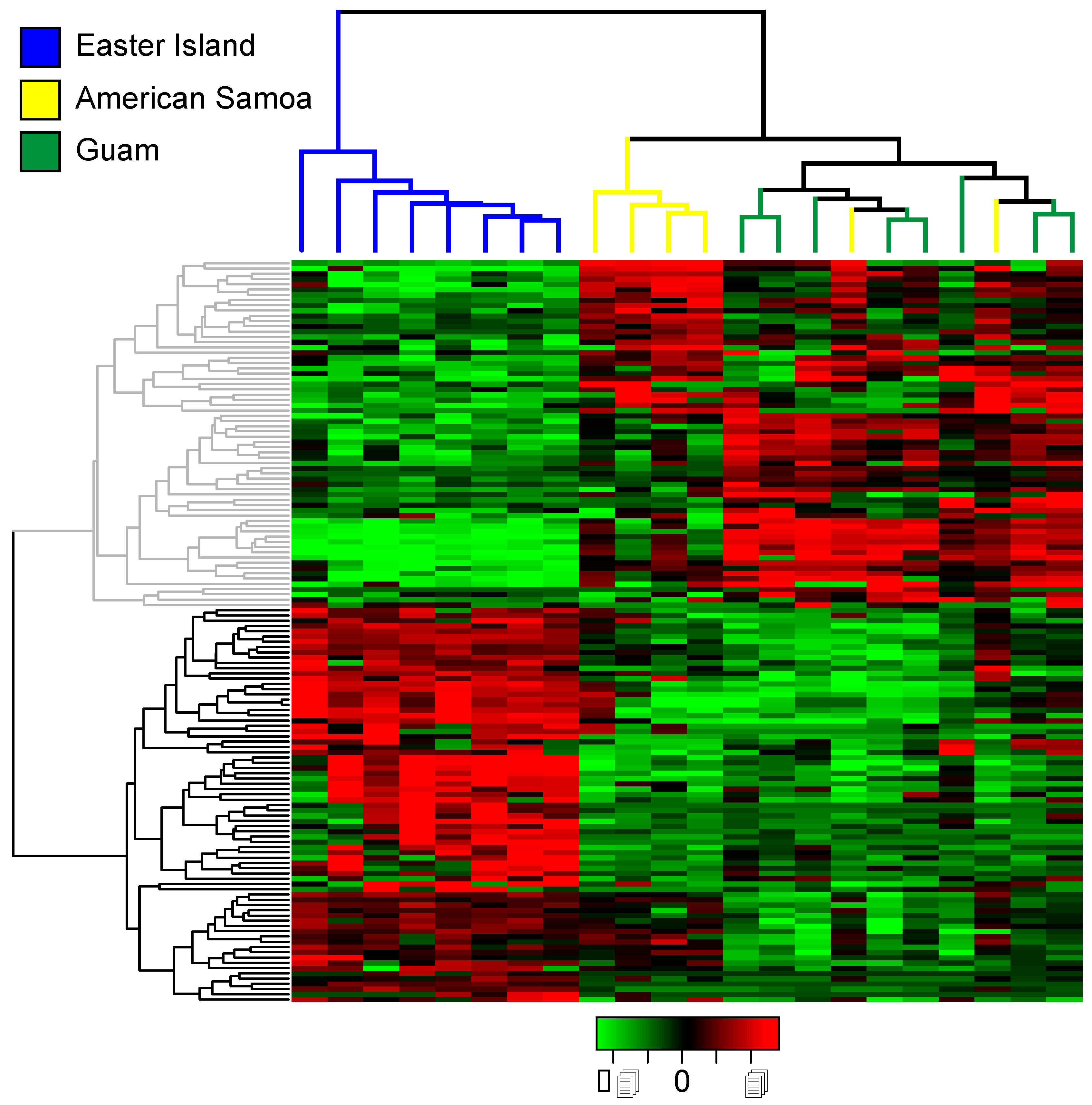
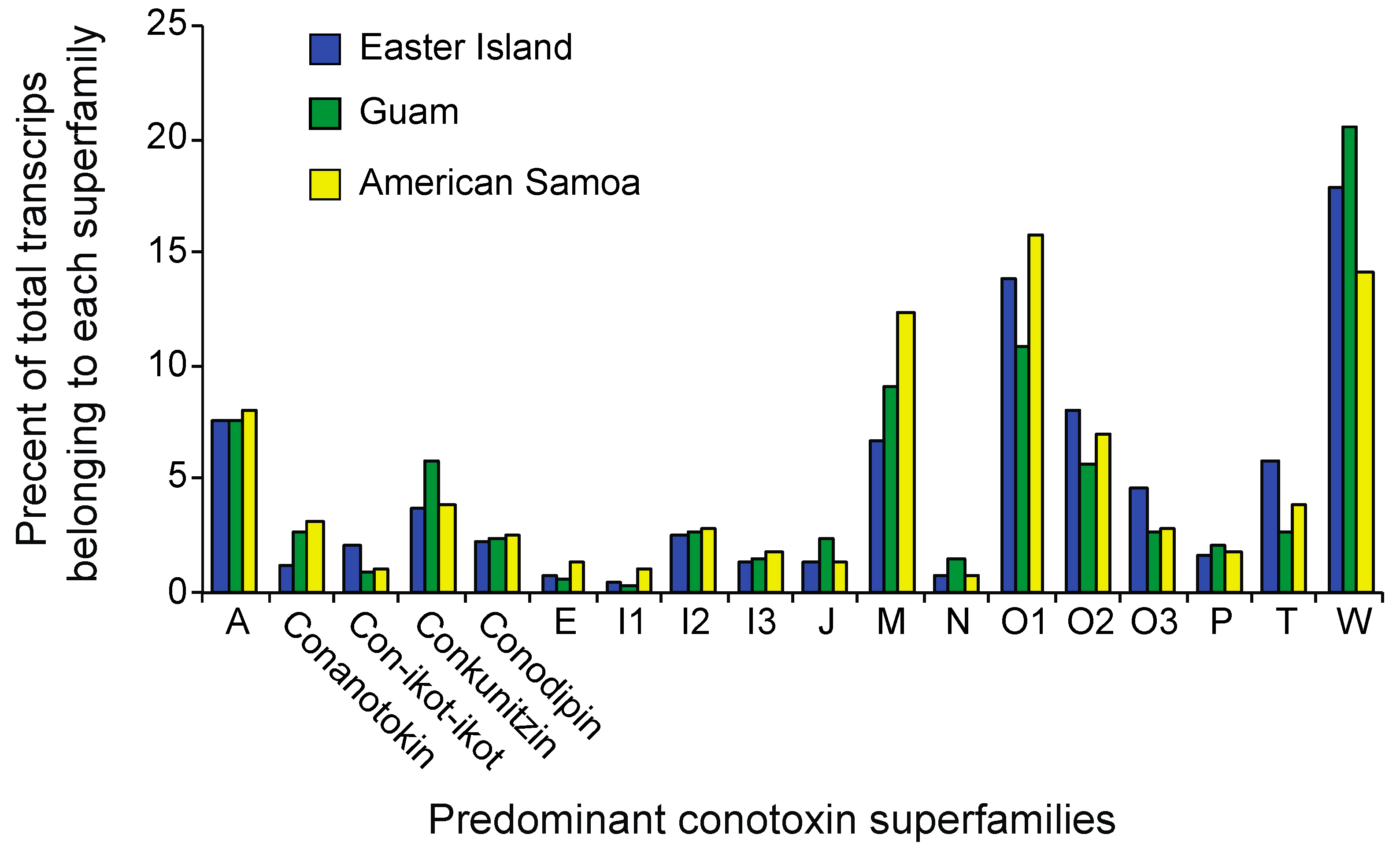
2.3. Differential Expression
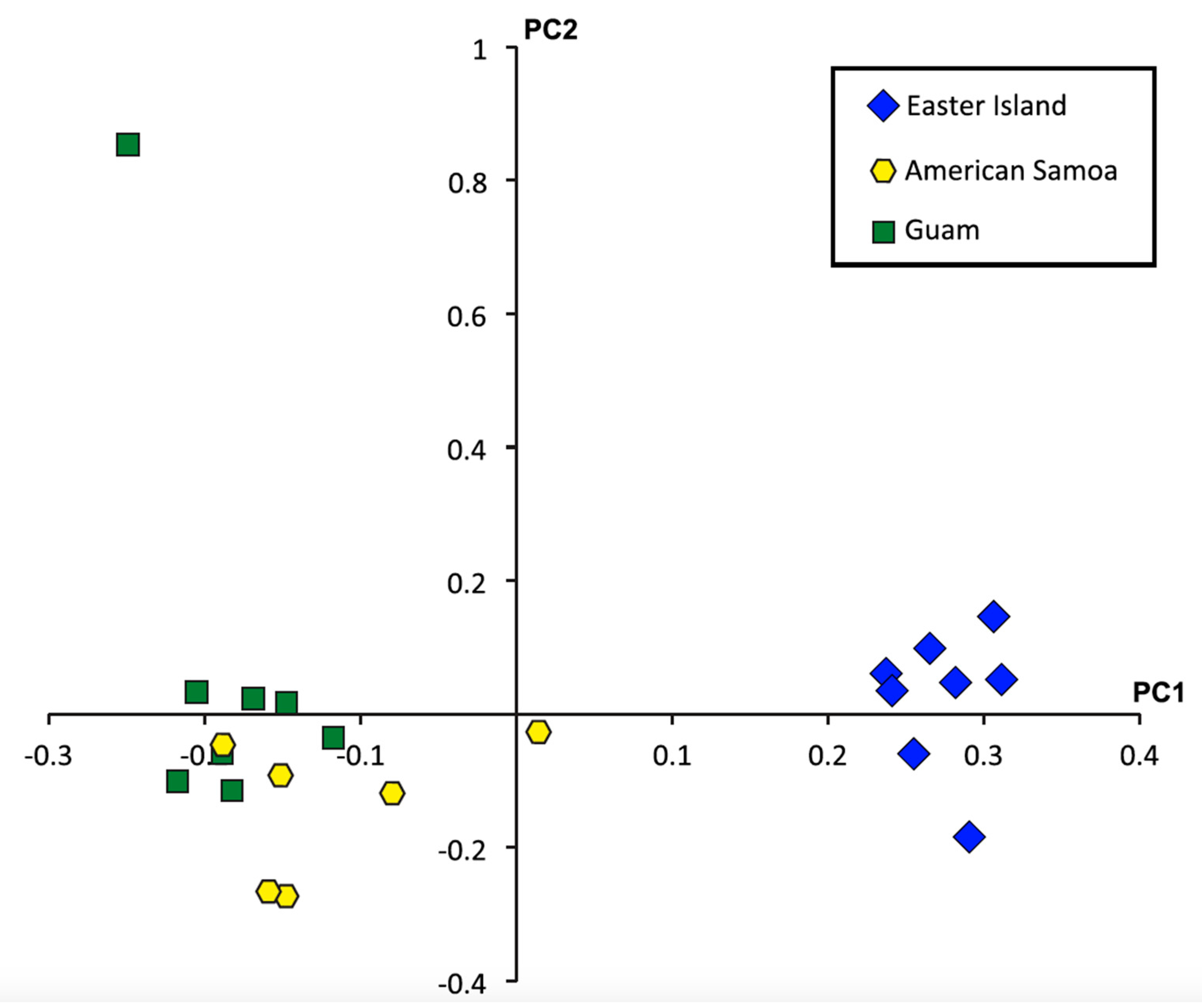
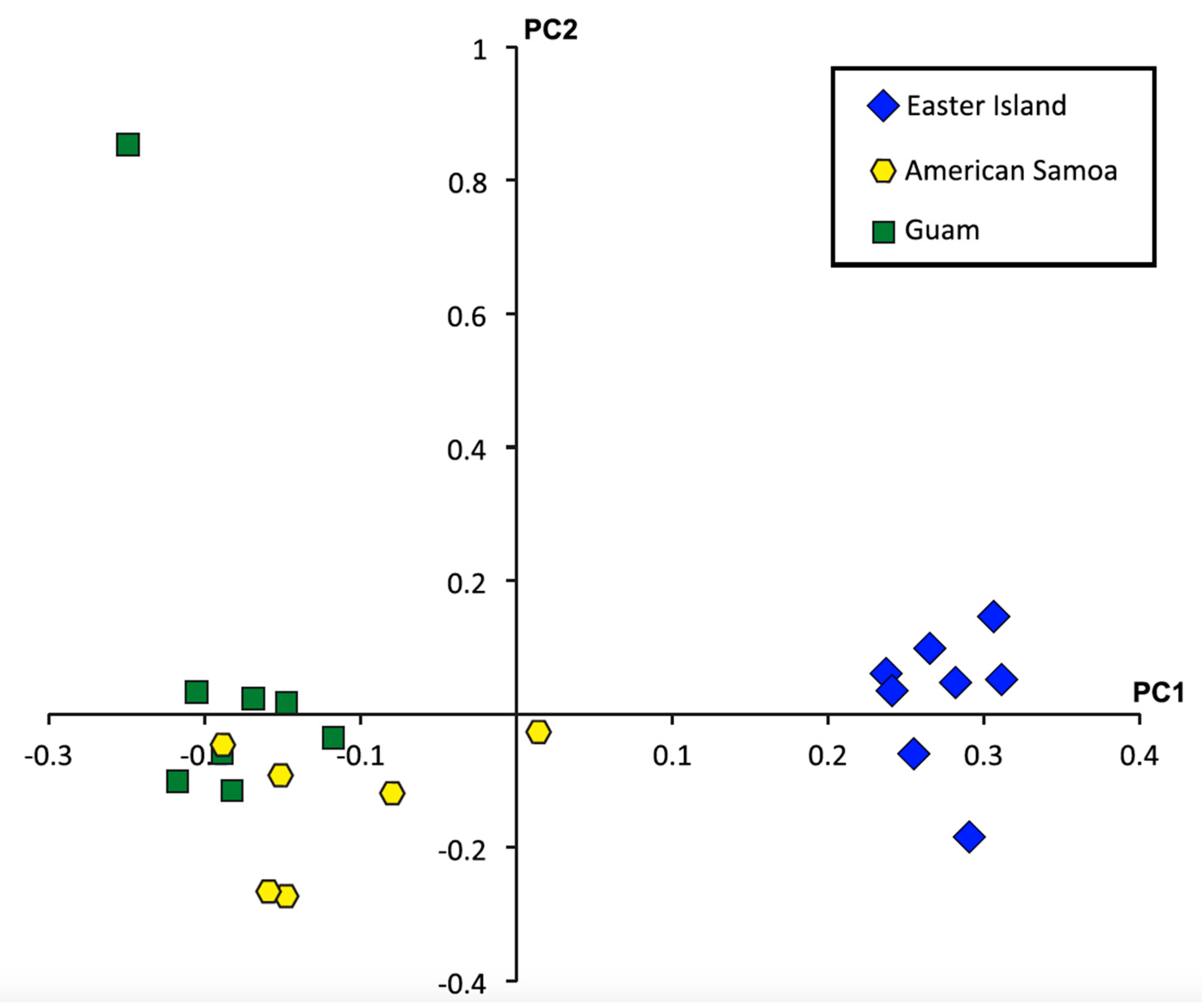
2.4. Single Nucleotide Polymorphisms, Diversity, and Population Structure
2.5. Data Accessibility
3. Discussion
3.1. Diets and Dietary Breadth
3.2. Mechanisms Contributing to the Differentiation of Venoms among Populations with Different Resource Use Patterns
4. Conclusions
5. Materials and Methods
5.1. Dietary Analyses
5.2. De novo Transcriptome Assembly and Annotation
5.3. Differential Expression
5.4. Single-Nucleotide Polymorphism (SNP) Identification, Diversity, Population Structure, and Outlier Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thompson, J.N. The Geographic Mosaic of Coevolution; University of Chicago: Chicago, IL, USA, 2005; ISBN 978-0-226-79762-5. [Google Scholar]
- Schluter, D. Evidence for ecological speciation and its alternative. Science 2009, 323, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.N. The coevolving web of life. Am. Nat. 2009, 173, 125–140. [Google Scholar] [CrossRef]
- Endler, J.A. A predator’s view of animal color patterns. Evol. Biol. 1978, 11, 319–364. [Google Scholar]
- Gervasi, D.D.L.; Schiestl, F.P. Real-time divergent evolution in plants driven by pollinators. Nat. Commun. 2017, 8, 14691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Duda, T.F.; Meyer, C.; Olivera, B.M.; Bouchet, P. One, four or 100 genera? A new classification of the cone snails. J. Molluscan Stud. 2015, 81, 1–23. [Google Scholar] [CrossRef]
- Olivera, B.M. Conus venom peptides: Reflections from the biology of clades and species. Annu. Rev. Ecol. Syst. 2002, 33, 25–47. [Google Scholar] [CrossRef]
- Duda, T.F., Jr. Differentiation of venoms of predatory marine gastropods: Divergence of orthologous toxin genes of closely related Conus species with different dietary specializations. J. Mol. Evol. 2008, 67, 315–321. [Google Scholar] [CrossRef]
- Duda, T.F., Jr.; Remigio, E.A. Variation and evolution of toxin gene expression patterns of six closely related venomous marine snails. Mol. Ecol. 2008, 17, 3018–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, T.F., Jr.; Palumbi, S.R. Molecular genetics of ecological diversification: Duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Proc. Natl. Acad. Sci. USA 1999, 96, 6820–6823. [Google Scholar] [CrossRef]
- Chang, D.; Duda, T.F., Jr. Application of community phylogenetic approaches to understand gene expression: Differential exploration of venom gene space in predatory marine gastropods. BMC Evol. Biol. 2014, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Duda, T.F., Jr. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, H.L.; Rossiter, W. Rapid evolution by positive selection and gene gain and loss: PLA2 venom genes in closely related Sistrurus rattlesnakes with divergent diets. J. Mol. Evol. 2008, 66, 151–166. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Wray, K.P.; Margres, M.J. The genesis of an exceptionally lethal venom in the timber rattlesnake (Crotalus horridus) revealed through comparative venom-gland transcriptomics. BMC Genom. 2013, 14, 394. [Google Scholar] [CrossRef] [PubMed]
- Dowell, N.L.; Giorgianni, M.W.; Kassner, V.A.; Selegue, J.E.; Sanchez, E.E.; Carroll, S.B. The deep origin and recent loss of venom toxin genes in rattlesnakes. Curr. Biol. 2016, 26, 2434–2445. [Google Scholar] [CrossRef]
- Kohn, A.J. The Ecology of Conus in Hawaii. Ecol. Monogr. 1959, 29, 47–90. [Google Scholar] [CrossRef]
- Kohn, A.J. Food Specialization in Conus in Hawaii and California. Ecology 1966, 47, 1041–1043. [Google Scholar] [CrossRef]
- Kohn, A.J. Abundance, diversity and resource use in an assemblage of Conus species in Enewetak Lagoon. Pac. Sci. 1980, 34, 359–369. [Google Scholar]
- Marsh, H. Observations on the food and feeding of some vermivorous Conus on the Great Barrier Reef. Veliger 1971, 14, 45–53. [Google Scholar]
- Kohn, A.J.; Nybakken, J.W. Ecology of Conus on eastern Indian Ocean fringing reefs: Diversity of species and resource utilization. Mar. Biol. 1975, 29, 211–234. [Google Scholar] [CrossRef]
- Leviten, P.J. The foraging strategy of vermivorous conid gastropods. Ecol. Monogr. 1976, 46, 157–178. [Google Scholar] [CrossRef]
- Reichelt, R.E.; Kohn, A.J. Feeding and distribution of predatory gastropods on some Great Barrier Reef platforms. In Proceedings of the Fifth International Coral Reef Congress, Tahiti, France, 27 May–1 June 1985; Volume 5, pp. 191–196. [Google Scholar]
- Kohn, A.J.; Almasi, K.N. Comparative ecology of a biographically heterogeneous Conus assemblage. In Proceedings of the Fifth International Marine Biological Workshop: The Marine Flora and Fauna of Rottnest Island, Western Australia; Wells, F.E., Walker, D.I., Kirkman, H., Lethbridge, R., Eds.; Western Australia Museum: Pertha, Australia, 1993; pp. 509–521. [Google Scholar]
- Kohn, A.J. Ecological shift and release in an isolated population: Conus miliaris at Easter Island. Ecol. Monogr. 1978, 48, 323–336. [Google Scholar] [CrossRef]
- Kohn, A.J. Maximal species richness in Conus: Diversity, diet and habitat on reefs of northeast Papua New Guinea. Coral Reefs 2001, 20, 25–38. [Google Scholar]
- Duda, T.F., Jr.; Lee, T. Isolation and population divergence of a widespread Indo-West Pacific marine gastropod at Easter Island. Mar. Biol. 2009, 156, 1193–1202. [Google Scholar] [CrossRef]
- Duda, T.F., Jr.; Lee, T. Ecological release and venom evolution of a predatory marine snail at Easter Island. PLoS ONE 2009, 4, e5558. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.J.; Perron, F.E. Life History and Biogeography: Patterns in Conus; Clarendon Press: Oxford, UK, 1994; ISBN 978-0-19-854080-9. [Google Scholar]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prashanth, J.R.; Dutertre, S.; Lewis, R.J. Revising the Role of Defense and Predation in Cone Snail Venom Evolution. In Evolution of Venomous Animals and Their Toxins; Malhotra, A., Ed.; Springer: Dordrecht, The Netherlands, 2017; pp. 105–123. ISBN 978-94-007-6457-6. [Google Scholar]
- De Wit, P.; Palumbi, S.R. Transcriptome-wide polymorphisms of red abalone (Haliotis rufescens) reveal patterns of gene flow and local adaptation. Mol. Ecol. 2013, 22, 2884–2897. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Gyoja, F.; Tanaka, M.; Ikuta, T.; Shoguchi, E.; Fujiwara, M.; Shinzato, C.; Hisata, K.; et al. Draft genome of the pearl oyster Pinctada fucata: A platform for understanding bivalve biology. DNA Res. 2012, 19, 117–130. [Google Scholar] [CrossRef]
- Bolnick, D.I.; Svanbäck, R.; Fordyce, J.A.; Yang, L.H.; Davis, J.M.; Hulsey, C.D.; Forister, M.L. The ecology of individuals: Incidence and implications of individual specialization. Am. Nat. 2003, 161, 1–28. [Google Scholar] [CrossRef]
- Bolnick, D.I.; Ingram, T.; Stutz, W.E.; Snowberg, L.K.; Lau, O.L.; Paull, J.S. Ecological release from interspecific competition leads to decoupled changes in population and individual niche width. Proc. R. Soc. B Biol. Sci. 2010, 277, 1789–1797. [Google Scholar] [CrossRef]
- Svanbäck, R.; Bolnick, D.I. Intraspecific competition drives increased resource use diversity within a natural population. Proc. R. Soc. B Biol. Sci. 2007, 274, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Araújo, M.S.; Martins, E.G.; Cruz, L.D.; Fernandes, F.R.; Linhares, A.X.; Dos Reis, S.F.; Guimarães, P.R. Nested diets: A novel pattern of individual-level resource use. Oikos 2010, 119, 81–88. [Google Scholar] [CrossRef]
- Kohn, A.J.; Lloyd, M.C. Polychaetes of truncated reef limestone substrates on eastern Indian Ocean coral reefs: Diversity, abundance, and taxonomy. Int. Rev. Gesamten Hydrobiol. Hydrogr. 1973, 58, 369–400. [Google Scholar] [CrossRef]
- Van Valen, L. Morphological variation and width of ecological niche. Am. Nat. 1965, 99, 377–390. [Google Scholar] [CrossRef]
- Roughgarden, J. Evolution of niche width. Am. Nat. 1972, 106, 683–718. [Google Scholar] [CrossRef]
- Levis, N.A.; Martin, R.A.; O’Donnell, K.A.; Pfennig, D.W. Intraspecific adaptive radiation: Competition, ecological opportunity, and phenotypic diversification within species. Evolution 2017, 71, 2496–2509. [Google Scholar] [CrossRef] [PubMed]
- Phuong, M.A.; Mahardika, G.N. Targeted sequencing of venom genes from cone snail genomes improves understanding of conotoxin molecular evolution. Mol. Biol. Evol. 2018, 35, 1210–1224. [Google Scholar] [CrossRef]
- Chang, D.; Olenzek, A.M.; Duda, T.F., Jr. Effects of geographical heterogeneity in species interactions on the evolution of venom genes. Proc. R. Soc. B Biol. Sci. 2015, 282, 20141984. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Duda, T.F., Jr. Age-related association of venom gene expression and diet of predatory gastropods. BMC Evol. Biol. 2016, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Phuong, M.A.; Mahardika, G.N.; Alfaro, M.E. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genom. 2016, 17, 401. [Google Scholar] [CrossRef]
- Romero, I.G.; Ruvinsky, I.; Gilad, Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012, 13, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Xing, S.; Zhu, C.; Liu, W.; Fan, Y.; Wang, Q.; Song, Z.; Yang, W.; Luo, F.; Shang, F.; et al. Population transcriptomics reveals a potentially positive role of expression diversity in adaptation. J. Integr. Plant Biol. 2015, 57, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Toews, D.P.L.; Campagna, L.; Taylor, S.A.; Balakrishnan, C.N.; Baldassarre, D.T.; Deane-Coe, P.E.; Harvey, M.G.; Hooper, D.M.; Irwin, D.E.; Judy, C.D.; et al. Genomic approaches to understanding population divergence and speciation in birds. Auk Ornithol. Adv. 2016, 133, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, M.; Ravindran, S.P.; Schwenk, K.; Cordellier, M. Population transcriptomics in Daphnia: The role of thermal selection. Mol. Ecol. 2018, 27, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Chang, D.; Lewis, B.D.; Lee, T. Geographic variation in venom allelic composition and diets of the widespread predatory marine gastropod Conus ebraeus. PLoS ONE 2009, 4, e6245. [Google Scholar] [CrossRef] [PubMed]
- Palumbi, S.R. Nucleic acids II: The polymerase chain reaction. In Molecular Systematics; Sinauer & Associates: Sunderland, MA, USA, 1996; pp. 205–247. [Google Scholar]
- Rambaut, A. Se-Al: Sequence Alignment Editor; University of Oxford: Oxford, UK, 1996. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.H. A study of summer foliage insect communities in the Great Smoky Mountains. Ecol. Monogr. 1952, 22, 1–44. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 2 June 2014).
- MacManes, M.D. On the optimal trimming of high-throughput mRNA sequence data. Front. Genet. 2014, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.T.; Howe, A.; Zhang, Q.; Pyrkosz, A.B.; Brom, T.H. A reference-free algorithm for computational normalization of shotgun sequencing data. ArXiv12034802 Q-Bio 2012. Available online: https://arxiv.org/abs/1203.4802 (accessed on 2 June 2014).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, Q.; Westermann, J.-C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 2 June 2014).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Introduction to the GATK Best Practices. Available online: https://software.broadinstitute.org/gatk/best-practices/ (accessed on 2 June 2014).
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genepop ’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Prey Item/Statistic | Easter Island | Guam | American Samoa |
|---|---|---|---|
| Eunicidae (total) | 76 | 59 | 21 |
| Eunicidae ‘X6’ | 40 | ||
| Eunicidae ‘X7’ | 10 | ||
| Eunicidae ‘X8’ | 2 | 4 | 13 |
| Eunicidae ‘X9’ | 13 | ||
| Eunicidae ‘X10’ | 5 | ||
| Eunicidae ‘X13’ | 19 | ||
| Eunicidae ‘X15’ | 2 | ||
| Eunicidae ‘X16’ | 4 | ||
| Eunicidae ‘X18’ | 4 | ||
| Palola A1 | 35 | ||
| Palola A3 | 5 | ||
| Onuphidae (‘O1’) | 4 | ||
| Lumbrinereidae (‘L1’) | 1 | ||
| Nereididae (‘N2’) | 8 | ||
| Capitellidae (‘C1’) | 1 | ||
| Number of prey items identified | 90 | 59 | 21 |
| Inferred number of species | 10 | 4 | 3 |
| Inferred number of families | 5 | 1 | 1 |
| Shannon diversity index of prey | 1.77 | 0.98 | 0.93 |
| Average genetic distances among prey | 0.321 | 0.114 | 0.161 |
| Statistic | C. miliaris | Easter Island | Guam | American Samoa |
|---|---|---|---|---|
| Number of PE reads | 349,293,902 | 131,525,916 | 130,566,380 | 87,201,606 |
| Total transcripts | 204,951 | 149,583 | 134,741 | 98,828 |
| Average transcript length (bp) | 624.2 | 560.4 | 550.5 | 428.9 |
| Longest transcript (bp) | 25,902 | 23,251 | 16,001 | 15,460 |
| N50 (bp) | 879 | 719 | 693 | 463 |
| Annotated transcripts (%) | 31,099 (15.2) | 23,166 (15.5) | 24,303 (16.3) | 17,956 (18.2) |
| Toxin-related transcripts | 516 | 435 | 340 | 284 |
| % of total reads mapped to toxin-related transcripts | 66 | 59 | 72 | 65 |
| Population | All SNPs | Non-Toxin-Related SNPs | Toxin-Related SNPs |
|---|---|---|---|
| Easter Island | 0.159 | 0.155 | 0.198 |
| Guam | 0.153 | 0.153 | 0.149 |
| American Samoa | 0.157 | 0.157 | 0.155 |
| Population | Easter Island | Guam | American Samoa |
|---|---|---|---|
| Easter Island | - | 0.121 | 0.143 |
| Guam | 0.069 | - | −0.012 |
| American Samoa | 0.054 | 0.001 | - |
| Population | Easter Island | Guam | American Samoa |
|---|---|---|---|
| Easter Island | - | 0.050 | 0.034 |
| Guam | 0.207 | - | 0.008 |
| American Samoa | 0.178 | 0.035 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weese, D.A.; Duda, T.F., Jr. Effects of Predator-Prey Interactions on Predator Traits: Differentiation of Diets and Venoms of a Marine Snail. Toxins 2019, 11, 299. https://doi.org/10.3390/toxins11050299
Weese DA, Duda TF Jr. Effects of Predator-Prey Interactions on Predator Traits: Differentiation of Diets and Venoms of a Marine Snail. Toxins. 2019; 11(5):299. https://doi.org/10.3390/toxins11050299
Chicago/Turabian StyleWeese, David A., and Thomas F. Duda, Jr. 2019. "Effects of Predator-Prey Interactions on Predator Traits: Differentiation of Diets and Venoms of a Marine Snail" Toxins 11, no. 5: 299. https://doi.org/10.3390/toxins11050299
APA StyleWeese, D. A., & Duda, T. F., Jr. (2019). Effects of Predator-Prey Interactions on Predator Traits: Differentiation of Diets and Venoms of a Marine Snail. Toxins, 11(5), 299. https://doi.org/10.3390/toxins11050299