Detection of Five Mycotoxins in Different Food Matrices in the Malaysian Market by Using Validated Liquid Chromatography Electrospray Ionization Triple Quadrupole Mass Spectrometry


Abstract
:1. Introduction
2. Results
2.1. Optimization of the MS/MS Conditions
2.2. Univariate Optimization
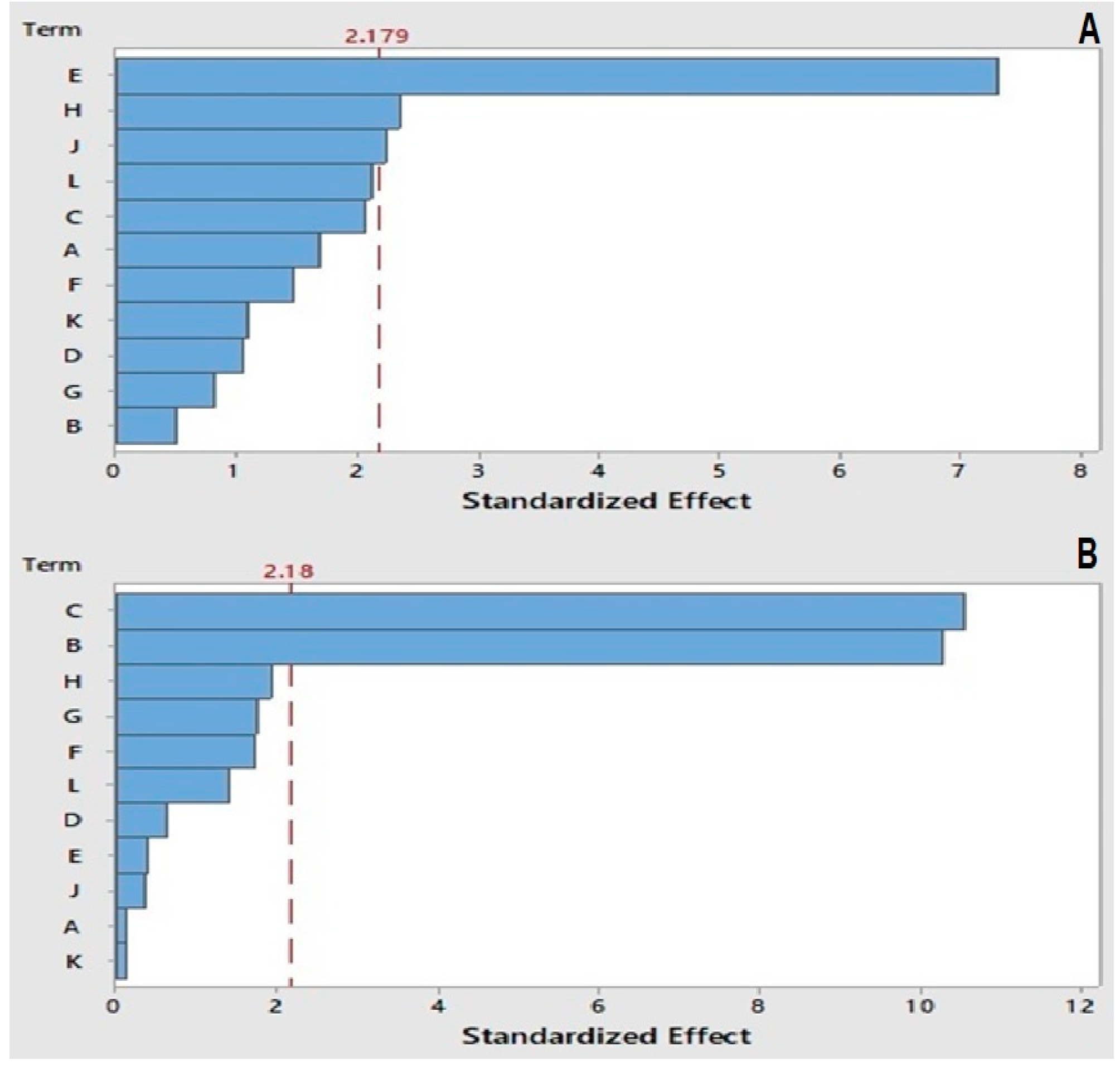
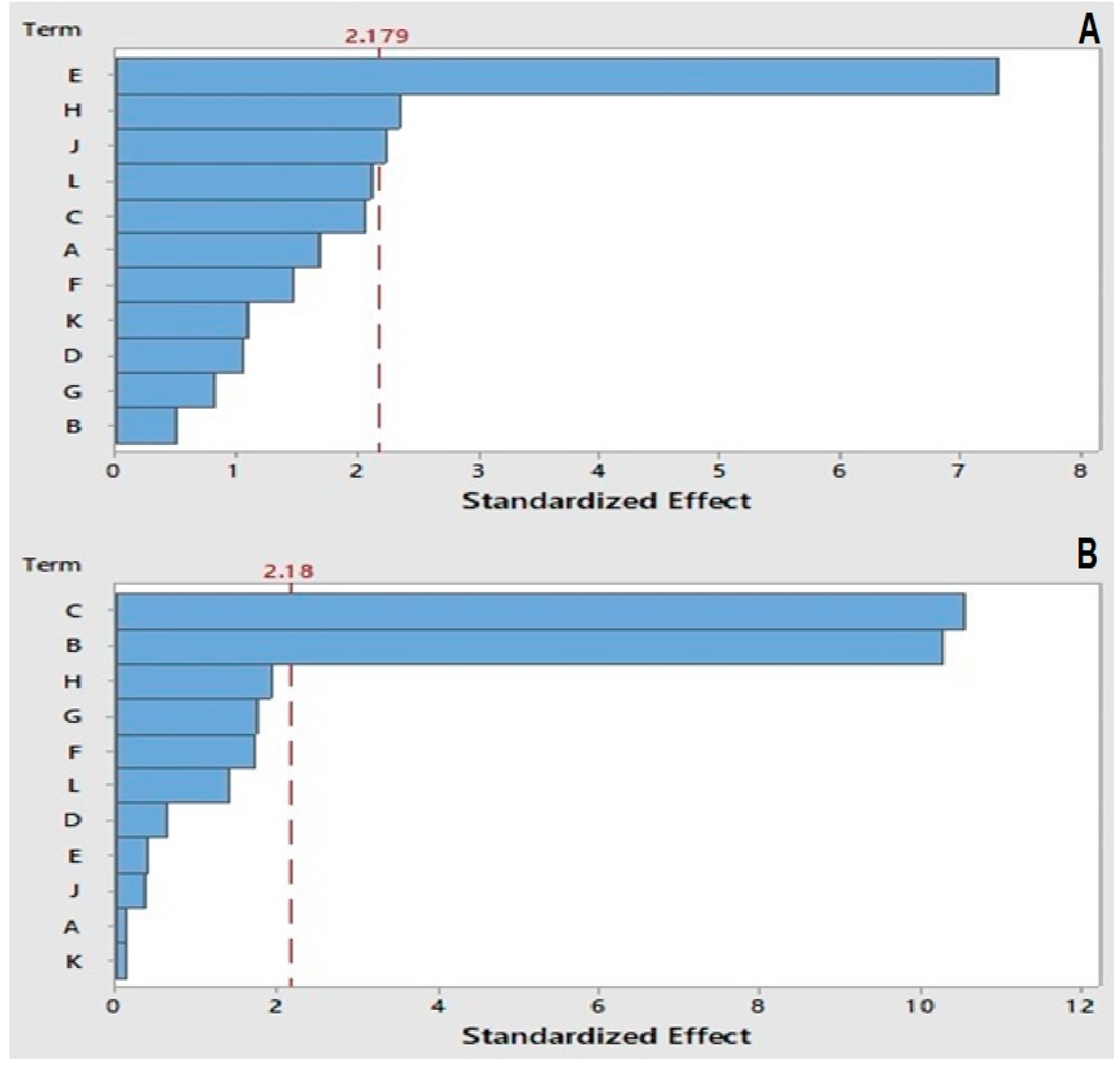
2.3. Plackett–Burman Design (PBD)
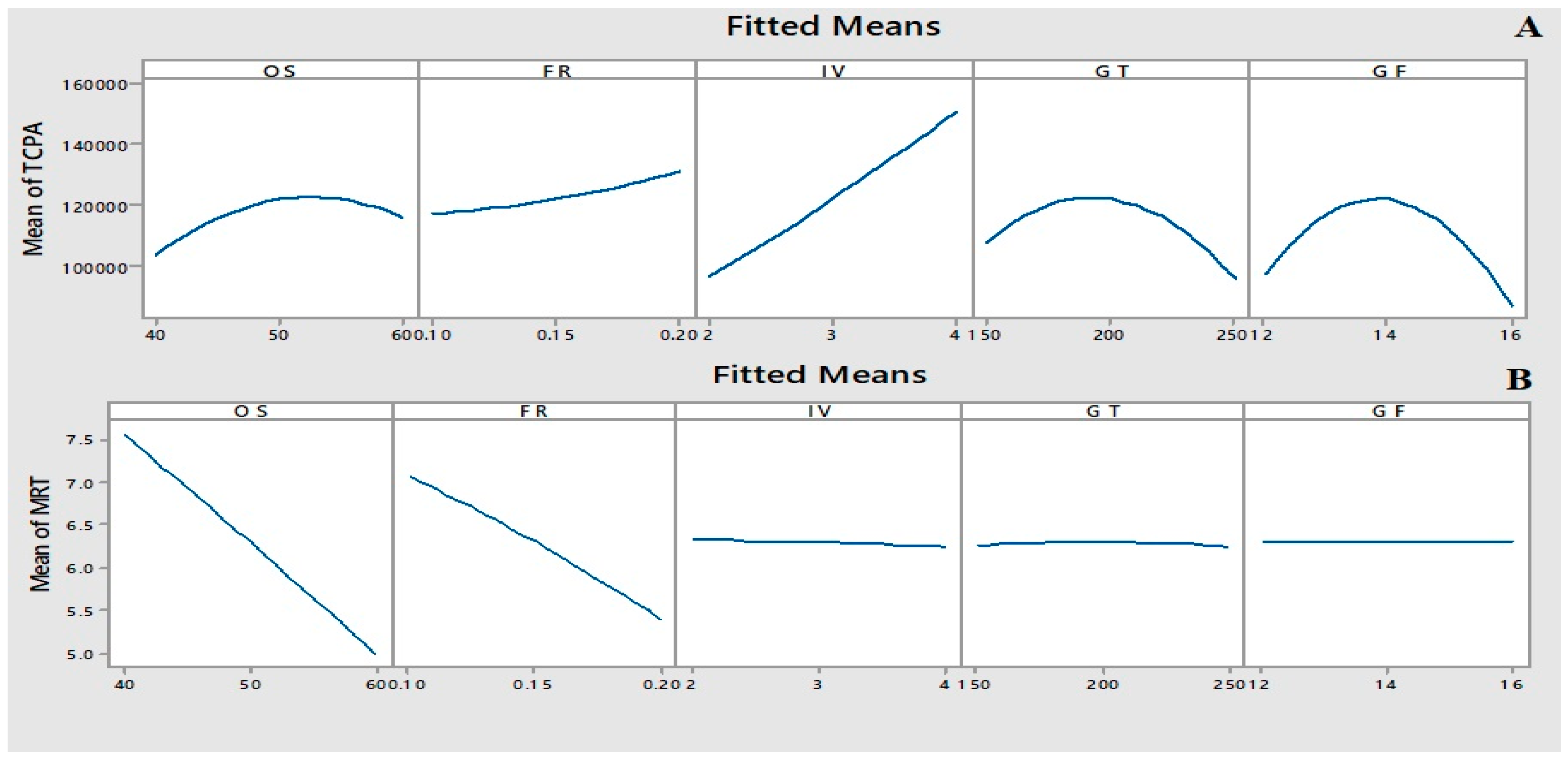
2.4. Box–Behnken Design (BBD)
2.5. Method Performance
2.6. Comparison of the Developed Method with Other Methods
2.7. Occurrence of Studied Mycotoxins in Real Food Samples
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Samples
4.3. LC-MS Instrumentation
4.4. Non- and Low-Fat Sample Preparation
4.5. High-Fat Sample Preparation
4.6. Optimization Method
4.7. Method Performance
4.7.1. Instrument Validation
4.7.2. Detection Method Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Logrieco, A.F.; Miller, J.D.; Eskola, M.; Krska, R.; Ayalew, A.; Bandyopadhyay, R.; Battilani, P.; Bhatnagar, D.; Chulze, S.; De Saeger, S. The Mycotox Charter: Increasing awareness of, and concerted action for, minimizing mycotoxin exposure worldwide. Toxins 2018, 10, 149. [Google Scholar] [CrossRef] [PubMed]
- Negash, D. A review of aflatoxin: Occurrence, prevention, and gaps in both food and feed safety. J. Appl. Microbiol. Res. 2018, 1, 35–43. [Google Scholar] [CrossRef]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens—The IARC Monographs classification. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Yusefi, J.; Valaee, M.; Nazari, F.; Maleki, J.; Mottaghianpour, E.; Khosrokhavar, R.; Hosseini, M.-J. Occurrence of ochratoxin a in grape juice of Iran. Iran. J. Pharm. Res. IJPR 2018, 17, 140. [Google Scholar] [PubMed]
- Ukwuru, M.; Ohaegbu, C.; Muritala, A. An overview of mycotoxin contamination of foods and feeds. J. Biochem. Microb. Toxicol. 2017, 1, 101. [Google Scholar]
- Udomkun, P.; Wiredu, A.N.; Nagle, M.; Müller, J.; Vanlauwe, B.; Bandyopadhyay, R. Innovative technologies to manage aflatoxins in foods and feeds and the profitability of application—A review. Food Control 2017, 76, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, A.; Solfrizzo, M.; Bibi, R.; Pecorelli, I. Development and validation of LC-MS/MS method for the determination of Ochratoxin A and its metabolite Ochratoxin α in poultry tissues and eggs. J. Environ. Sci. Health Part B 2018, 53, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Alshannaq, A.; Yu, J.-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed]
- De Santis, B.; Debegnach, F.; Gregori, E.; Russo, S.; Marchegiani, F.; Moracci, G.; Brera, C. Development of a LC-MS/MS Method for the multi-mycotoxin determination in composite cereal-based samples. Toxins 2017, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Bai, B.; Jin, P.; Fan, K.; Guo, W.; Zhao, Z.; Han, Z. Development and validation of an ultra-high performance liquid chromatography-tandem mass spectrometry method for simultaneous determination of four type B trichothecenes and masked deoxynivalenol in various feed products. Molecules 2016, 21, 747. [Google Scholar] [CrossRef] [PubMed]
- Sargent, M.; Committee, A.M. Guide to Achieving Reliable Quantitative LC-MS Measurements; RSC Analytical Methods Committee: Middlesex, UK, 2013. [Google Scholar]
- Tahboub, Y.R. Chromatographic behavior of co-eluted plasma compounds and effect on screening of drugs by APCI-LC–MS (/MS): Applications to selected cardiovascular drugs. J. Pharm. Anal. 2014, 4, 384–391. [Google Scholar] [CrossRef]
- Szerkus, B.; Mpanga, A.; Kaliszan, R.; Siluk, D. Optimization of the electrospray ionization source with the use of the design of experiments approach for the LC–MS-MS determination of selected metabolites in human urine. Spectroscopy 2016, 14, 8–16. [Google Scholar]
- Low, K.H.; Zain, S.M.; Abas, M.R. Evaluation of microwave-assisted digestion condition for the determination of metals in fish samples by inductively coupled plasma mass spectrometry using experimental designs. Int. J. Environ. Anal. Chem. 2012, 92, 1161–1175. [Google Scholar] [CrossRef]
- Low, K.; Zain, S.M.; Abas, M.R.; Mohd, A. Evaluation of Closed Vessel Microwave Digestion of Fish Muscle with Various Solvent Combinations Using Fractional Factorial Design. ASM Sci. J. 2009, 3, 71–76. [Google Scholar]
- Anderson, M.J.; Whitcomb, P.J. RSM Simplified: Optimizing Processes Using Response Surface Methods for Design of Experiments; Productivity Press: New York, NY, USA, 2016. [Google Scholar]
- Dos Santos Depoi, F.; Bentlin, F.R.; Ferrao, M.F.; Pozebon, D. Multivariate optimization for cloud point extraction and determination of lanthanides. Anal. Methods 2012, 4, 2809–2814. [Google Scholar] [CrossRef]
- Karapinar, I.; Ertaş, F.N.; Şahintürk, B.; Aftafa, C.; Kiliç, E. LC-MS/MS signal enhancement for estrogenic hormones in water samples using experimental design. RSC Adv. 2016, 6, 39188–39197. [Google Scholar] [CrossRef]
- Székely, G.; Henriques, B.; Gil, M.; Alvarez, C. Experimental design for the optimization and robustness testing of a liquid chromatography tandem mass spectrometry method for the trace analysis of the potentially genotoxic 1, 3-diisopropylurea. Drug Test. Anal. 2014, 6, 898–908. [Google Scholar] [CrossRef]
- Hasnain, M.S.; Rao, S.; Singh, M.K.; Vig, N.; Gupta, A.; Ansari, A.; Sen, P.; Joshi, P.; Ansari, S.A. Development and validation of LC–MS/MS method for the quantitation of lenalidomide in human plasma using Box–Behnken experimental design. Analyst 2013, 138, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.L.T.; Sanson, A.L.; de Vasconcelos Quaresma, A.; de Paiva Gomes, R.; da Silva, G.A.; Afonso, R.J.d.C.F. Chemometric approach to optimize the operational parameters of ESI for the determination of contaminants of emerging concern in aqueous matrices by LC-IT-TOF-HRMS. Microchem. J. 2014, 117, 242–249. [Google Scholar] [CrossRef]
- González-Jartín, J.M.; Alfonso, A.; Rodríguez, I.; Sainz, M.J.; Vieytes, M.R.; Botana, L.M. A QuEChERS based extraction procedure coupled to UPLC-MS/MS detection for mycotoxins analysis in beer. Food Chem. 2019, 275, 703–710. [Google Scholar] [CrossRef]
- Cao, X.; Li, X.; Li, J.; Niu, Y.; Shi, L.; Fang, Z.; Zhang, T.; Ding, H. Quantitative determination of carcinogenic mycotoxins in human and animal biological matrices and animal-derived foods using multi-mycotoxin and analyte-specific high performance liquid chromatography-tandem mass spectrometric methods. J. Chromatogr. B 2018, 1073, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, X.; Han, S.-Y.; Li, M.; Ma, T.-Z.; Sheng, W.-J.; Zhu, X. Simultaneous Analysis of 20 Mycotoxins in Grapes and Wines from Hexi Corridor Region (China): Based on a QuEChERS–UHPLC–MS/MS Method. Molecules 2018, 23, 1926. [Google Scholar] [CrossRef]
- Rejczak, T.; Tuzimski, T. A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem. 2015, 13. [Google Scholar] [CrossRef]
- Ferreira, S.L.; Caires, A.O.; Borges, T.d.S.; Lima, A.M.; Silva, L.O.; dos Santos, W.N. Robustness evaluation in analytical methods optimized using experimental designs. Microchem. J. 2017, 131, 163–169. [Google Scholar] [CrossRef]
- Abdulra’uf, L.B.; Tan, G.H. Chemometric approach to the optimization of HS-SPME/GC–MS for the determination of multiclass pesticide residues in fruits and vegetables. Food Chem. 2015, 177, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Stalikas, C.; Fiamegos, Y.; Sakkas, V.; Albanis, T. Developments on chemometric approaches to optimize and evaluate microextraction. J. Chromatogr. A 2009, 1216, 175–189. [Google Scholar] [CrossRef]
- Ekpenyong, M.G.; Antai, S.P.; Asitok, A.D.; Ekpo, B.O. Plackett-Burman Design and Response Surface Optimization of Medium Trace Nutrients for Glycolipopeptide Biosurfactant Production. Iran. Biomed. J. 2017, 21, 249. [Google Scholar] [CrossRef]
- Asati, A.; Satyanarayana, G.; Patel, D.K. Vortex-assisted surfactant-enhanced emulsification microextraction combined with LC–MS/MS for the determination of glucocorticoids in water with the aid of experimental design. Anal. Bioanal. Chem. 2017, 409, 2905–2918. [Google Scholar] [CrossRef] [PubMed]
- Hair, J.F., Jr.; Hult, G.T.M.; Ringle, C.; Sarstedt, M. A Primer on Partial Least Squares Structural Equation Modeling (PLS-SEM); Sage Publications: Los Angeles, CA, USA, 2016. [Google Scholar]
- Ediage, E.N.; Van Poucke, C.; De Saeger, S. A multi-analyte LC–MS/MS method for the analysis of 23 mycotoxins in different sorghum varieties: The forgotten sample matrix. Food Chem. 2015, 177, 397–404. [Google Scholar] [CrossRef]
- European Commission (EC). 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- Lehotay, S.J.; Mastovska, K.; Lightfield, A.R.; Gates, R.A. Multi-analyst, multi-matrix performance of the QuEChERS approach for pesticide residues in foods and feeds using HPLC/MS/MS analysis with different calibration techniques. J. AOAC Int. 2010, 93, 355–367. [Google Scholar]
- Yarita, T.; Aoyagi, Y.; Otake, T. Evaluation of the impact of matrix effect on quantification of pesticides in foods by gas chromatography–mass spectrometry using isotope-labeled internal standards. J. Chromatogr. A 2015, 1396, 109–116. [Google Scholar] [CrossRef]
- Cunha, S.C.; Sá, S.V.; Fernandes, J.O. Multiple mycotoxin analysis in nut products: Occurrence and risk characterization. Food Chem. Toxicol. 2018, 114, 260–269. [Google Scholar] [CrossRef]
- Liu, Y.; Han, S.; Lu, M.; Wang, P.; Han, J.; Wang, J. Modified QuEChERS method combined with ultra-high performance liquid chromatography tandem mass spectrometry for the simultaneous determination of 26 mycotoxins in sesame butter. J. Chromatogr. B 2014, 970, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-D.; Suh, J.H.; Feng, S.; Eom, T.; Kim, J.; Hyun, S.M.; Kim, J.; Wang, Y.; Han, S.B. Comprehensive analysis of multi-class mycotoxins in twenty different species of functional and medicinal herbs using liquid chromatography–tandem mass spectrometry. Food Control 2019, 96, 517–526. [Google Scholar] [CrossRef]
- European Commission (EC). 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, 364, 5–24. [Google Scholar]
- Hong, L.S.; Yusof, N.I.M.; Ling, H.M. Determination of aflatoxins B1 and B2 in peanuts and corn based products. Sains Malays. 2010, 39, 731–735. [Google Scholar]
- Norlia, M.; Nor-Khaizura, M.; Selamat, J.; Abu Bakar, F.; Radu, S.; Chin, C.K. Evaluation of aflatoxin and Aspergillus sp. contamination in raw peanuts and peanut-based products along this supply chain in Malaysia. Food Addit. Contam. Part A 2018, 35, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.R.; Chye, F.Y.; Hamid, A.A.; Yatim, A.M. The occurrence of aflatoxins in raw shelled peanut samples from three districts of Perak, Malaysia. Electron. J. Environ. Agric. Food Chem. 2007, 6, 2045–2052. [Google Scholar]
- Norlia, M.; Jinap, S.; Nor-Khaizura, M.; Son, R.; Chin, C. Polyphasic approach to the identification and characterization of aflatoxigenic strains of Aspergillus section Flavi isolated from peanuts and peanut-based products marketed in Malaysia. Int. J. Food Microbiol. 2018, 282, 9–15. [Google Scholar] [CrossRef]
- Mohd-Redzwan, S.; Jamaluddin, R.; Mutalib, A.; Sokhini, M.; Ahmad, Z. A mini review on aflatoxin exposure in Malaysia: Past, present and future. Front. Microbiol. 2013, 4, 334. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Stacy, S.L.; Kensler, T.W. Global risk assessment of aflatoxins in maize and peanuts: Are regulatory standards adequately protective? Toxicol. Sci. 2013, 135, 251–259. [Google Scholar] [CrossRef]
- Jeswal, P.; Kumar, D. Mycobiota and natural incidence of aflatoxins, ochratoxin A, and citrinin in Indian spices confirmed by LC-MS/MS. Int. J. Microbiol. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Vaclavik, L.; Zachariasova, M.; Hrbek, V.; Hajslova, J. Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta 2010, 82, 1950–1957. [Google Scholar] [CrossRef]
- Wells, G.; Prest, H.; Russ IV, C.W. Why Use Signal-To-Noise As a Measure of MS Performance When It Is Often Meaningless? 2011. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/5990-8341EN.pdf (accessed on 28 February 2019).
- Kafeenah, H.I.; Osman, R.; Abu Bakar, N.K. UPLC-MS/MS screening method for simultaneous identification and characterisation of acidic and basic pharmaceuticals. Int. J. Environ. Anal. Chem. 2018, 98, 1207–1225. [Google Scholar] [CrossRef]
- Gjelstad, A.; Pedersen-Bjergaard, S. Perspective: Hollow fibre liquid-phase microextraction–principles, performance, applicability, and future directions. Sci. Chromatogr. 2013, 5, 181–189. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Mycotoxin | Molecular Formula | m/z | [M + H]+ | Product Ion (m/z) | Collision Energy |
|---|---|---|---|---|---|
| AFB1 | C17H12O6 | 312.0634 | 313.1 | 241.1 | 41 |
| 285.2 | 25 | ||||
| AFB2 | C17H14O6 | 314.0790 | 315.1 | 259.1 | 29 |
| 287.2 | 29 | ||||
| AFG1 | C17H12O7 | 328.0583 | 329.1 | 243.1 | 29 |
| 215.2 | 37 | ||||
| AFG2 | C17H14O7 | 330.0740 | 331.1 | 313.1 | 25 |
| 245.0 | 29 | ||||
| OTA | C20H18ClNO6 | 403.0823 | 404.0 | 221.0 | 38 |
| 239.0 | 26 |
| Factors | Optimized Univariate Parameter | Optimized Multivariate Parameter | PBD Lower Level | PBD Upper Level | BBD Lower Level | BBD Upper Level |
|---|---|---|---|---|---|---|
| Mobile phase additive | Ammonium formate & formic acid | n.a. | n.a. | n.a. | n.a. | n.a. |
| Mobile phase pH | 3 | 3 | 3 | 7 | n.a. | n.a. |
| Organic solvent (%) | 50 | 60 | 30 | 60 | 40 | 60 |
| Flow rate (mL/min) | 0.15 | 0.2 | 0.10 | 0.20 | 0.10 | 0.20 |
| Column temperature (°C) | 30 | 30 | 25 | 35 | n.a. | n.a. |
| Injection volume (L) | 3 | 4 | 2 | 5 | 2 | 4 |
| Sheath gas flow rate (L/min) | 11 | 11 | 9 | 12 | n.a. | n.a. |
| Sheath gas temperature (°C) | 250 | 250 | 150 | 250 | n.a. | n.a. |
| Gas flow rate (L/min) | 12 | 14 | 12 | 18 | 12 | 16 |
| Gas temperature (°C) | 200 | 170 | 150 | 250 | 150 | 250 |
| Nebulizer pressure (psi) | 25 | 25 | 20 | 35 | n.a. | n.a. |
| Collision energy (eV) | n.a. | n.a. | 25 | 40 | n.a. | n.a. |
| Source | DF | TCPA | MRT | ||
|---|---|---|---|---|---|
| F-Value | p-Value | F-Value | p-Value | ||
| Model | 20 | 2.53 | 0.015 | 133.30 | 0.000 |
| Blocks | 1 | 4.62 | 0.042 | 7.31 | 0.012 |
| Linear | 5 | 4.18 | 0.007 | 547.40 | 0.000 |
| OS | 1 | 0.91 | 0.350 | 1927.53 | 0.000 |
| FR | 1 | 1.10 | 0.305 | 807.80 | 0.000 |
| IV | 1 | 17.33 | 0.000 | 1.60 | 0.219 |
| GT | 1 | 0.87 | 0.361 | 0.06 | 0.806 |
| GF | 1 | 0.70 | 0.412 | 0.01 | 0.908 |
| Square | 5 | 3.61 | 0.014 | 0.82 | 0.550 |
| OS*OS | 1 | 1.85 | 0.186 | 0.44 | 0.513 |
| FR*FR | 1 | 0.07 | 0.798 | 3.02 | 0.095 |
| IV*IV | 1 | 0.04 | 0.848 | 0.19 | 0.664 |
| GT*GT | 1 | 5.20 | 0.032 | 1.18 | 0.287 |
| GF*GF | 1 | 11.66 | 0.002 | 0.02 | 0.889 |
| 2-Way Interaction | 10 | 0.96 | 0.497 | 5.10 | 0.001 |
| OS*FR | 1 | 0.35 | 0.560 | 45.54 | 0.000 |
| OS*IV | 1 | 4.62 | 0.042 | 0.95 | 0.338 |
| OS*GT | 1 | 0.02 | 0.882 | 0.43 | 0.517 |
| OS*GF | 1 | 0.08 | 0.781 | 1.26 | 0.273 |
| FR*IV | 1 | 1.00 | 0.326 | 0.75 | 0.395 |
| FR*GT | 1 | 0.00 | 0.998 | 1.72 | 0.202 |
| FR*GF | 1 | 2.39 | 0.135 | 0.00 | 0.983 |
| IV*GT | 1 | 0.30 | 0.588 | 0.12 | 0.737 |
| IV*GF | 1 | 0.27 | 0.606 | 0.01 | 0.910 |
| GT*GF | 1 | 0.60 | 0.445 | 0.21 | 0.648 |
| Lack-of-Fit | 20 | 0.95 | 0.593 | 0.57 | 0.825 |
| Mycotoxins | AFB1 | AFB2 | AFG1 | AFG2 | OTA | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Range (µg/L) | 0.018–50 | 0.012–15 | 0.018–50 | 0.012–15 | 0.02–50 | |||||
| a | b | a | b | a | b | a | b | a | b | |
| R2 | 0.9999 | 0.9998 | 0.9994 | 0.9992 | 0.9997 | 0.9995 | 0.9999 | 0.9998 | 0.9998 | 0.9993 |
| IDL (ng) | 1.75 | 2.35 | 2.93 | 3.01 | 2.60 | 3.22 | 3.12 | 3.61 | 1.41 | 2.48 |
| Intra-day Precision (RSD%) | 1.32 | 1.67 | 0.64 | 1.84 | 0.81 | 1.23 | 1.17 | 1.25 | 2.75 | 3.67 |
| Inter-day Precision (RSD%) | 2.78 | 2.95 | 1.63 | 1.70 | 0.97 | 1.80 | 3.6 | 3.11 | 3.3 | 3.89 |
| Mycotoxins | Apple Juice | Raisin | Wheat Flour | Peanut | Spice Mixture | |
|---|---|---|---|---|---|---|
| AFB1 | Range | 1–30 | 1–30 | 1–30 | 1–30 | 1–30 |
| R2 | 0.9991 | 0.9994 | 0.9993 | 0.9991 | 0.9989 | |
| LOD | 0.05 | 0.06 | 0.05 | 0.08 | 0.08 | |
| LOQ | 0.08 | 0.08 | 0.08 | 0.13 | 0.13 | |
| AFB2 | Range | 0.3–10 | 0.3–10 | 0.3–10 | 0.3–10 | 0.3–10 |
| R2 | 0.9990 | 0.9989 | 0.9990 | 0.9988 | 0.9987 | |
| LOD | 0.06 | 0.05 | 0.05 | 0.08 | 0.08 | |
| LOQ | 0.09 | 0.09 | 0.08 | 0.10 | 0.10 | |
| AFG1 | Range | 1–30 | 1–30 | 1–30 | 1–30 | 1–30 |
| R2 | 0.9992 | 0.9992 | 0.9991 | 0.9990 | 0.9989 | |
| LOD | 0.08 | 0.075 | 0.08 | 0.08 | 0.08 | |
| LOQ | 0.13 | 0.13 | 0.13 | 0.13 | 0.13 | |
| AFG2 | Range | 0.3–10 | 0.3–10 | 0.3–10 | 0.3–10 | 0.3–10 |
| R2 | 0.9989 | 0.9986 | 0.9987 | 0.9986 | 0.9984 | |
| LOD | 0.06 | 0.05 | 0.05 | 0.08 | 0.08 | |
| LOQ | 0.09 | 0.09 | 0.08 | 0.10 | 0.10 | |
| OTA | Range | 1–30 | 1–30 | 1–30 | 0.1–30 | 1–30 |
| R2 | 0.9991 | 0.9991 | 0.9989 | 0.9968 | 0.9967 | |
| LOD | 0.07 | 0.07 | 0.08 | 0.09 | 0.1 | |
| LOQ | 0.10 | 0.10 | 0.20 | 0.20 | 0.30 | |
| Mycotoxins | Conc a | Apple juice | Raisin | Wheat Flour | |||||||||||
| RE | Intra | Inter | RE | Intra | Inter | RE | Intra | Inter | |||||||
| AFB1 | 5 | 97.40 | 1.50 | 2.12 | 100.15 | 0.69 | 0.82 | 99.62 | 3.00 | 2.49 | |||||
| 10 | 99.92 | 1.38 | 4.01 | 99.45 | 1.16 | 1.28 | 100.70 | 0.46 | 0.43 | ||||||
| 30 | 99.98 | 0.21 | 0.23 | 99.64 | 3.43 | 4.86 | 99.80 | 0.54 | 2.30 | ||||||
| AFB2 | 1.5 | 100.72 | 0.96 | 3.38 | 99.60 | 1.34 | 1.10 | 97.33 | 1.22 | 2.48 | |||||
| 3 | 99.19 | 1.06 | 1.33 | 100.11 | 2.30 | 3.57 | 99.53 | 1.09 | 1.16 | ||||||
| 10 | 100.27 | 1.94 | 3.27 | 99.28 | 1.99 | 3.31 | 98.40 | 1.30 | 2.57 | ||||||
| AFG1 | 5 | 96.75 | 1.17 | 2.25 | 98.80 | 1.61 | 1.01 | 99.07 | 1.17 | 3.46 | |||||
| 10 | 100.87 | 1.75 | 4.21 | 99.45 | 1.07 | 5.60 | 100.1 | 0.65 | 6.62 | ||||||
| 30 | 99.66 | 2.84 | 1.55 | 98.78 | 2.68 | 2.98 | 101.45 | 2.82 | 2.30 | ||||||
| AFG2 | 1.5 | 96.75 | 1.18 | 1.90 | 98.80 | 1.60 | 5.47 | 88.80 | 3.01 | 6.18 | |||||
| 3 | 97.45 | 1.10 | 1.77 | 99.45 | 1.06 | 1.30 | 96.63 | 0.65 | 3.65 | ||||||
| 10 | 99.09 | 0.70 | 0.75 | 97.73 | 0.60 | 2.94 | 97.83 | 4.04 | 2.77 | ||||||
| OTA | 5 | 96.75 | 1.17 | 1.92 | 90.70 | 1.61 | 1.01 | 94.29 | 6.37 | 4.04 | |||||
| 10 | 99.56 | 0.48 | 0.54 | 97.21 | 1.03 | 1.40 | 100.7 | 0.66 | 6.48 | ||||||
| 30 | 98.04 | 0.55 | 1.35 | 98.28 | 0.12 | 0.88 | 97.93 | 0.55 | 1.35 | ||||||
| Conc a | Peanut | Spice Mixture | |||||||||||||
| RE | Intra | Inter | RE | Intra | Inter | ||||||||||
| AFB1 | 5 | 98.92 | 0.61 | 1.11 | 93.80 | 1.65 | 3.37 | ||||||||
| 10 | 99.88 | 1.06 | 1.93 | 97.81 | 2.33 | 1.63 | |||||||||
| 30 | 98.88 | 0.60 | 3.29 | 99.73 | 0.89 | 5.55 | |||||||||
| AFB2 | 1.5 | 92.58 | 0.80 | 4.24 | 90.43 | 3.16 | 7.20 | ||||||||
| 3 | 96.31 | 1.70 | 2.30 | 96.81 | 2.33 | 6.81 | |||||||||
| 10 | 99.93 | 0.54 | 2.65 | 98.54 | 6.29 | 4.20 | |||||||||
| AFG1 | 5 | 95.06 | 5.48 | 2.28 | 99.82 | 1.17 | 1.63 | ||||||||
| 10 | 101.67 | 1.06 | 10.10 | 97.81 | 2.33 | 8.04 | |||||||||
| 30 | 97.60 | 1.85 | 6.27 | 99.83 | 0.61 | 3.34 | |||||||||
| AFG2 | 1.5 | 88.03 | 1.17 | 7.10 | 84.52 | 1.18 | 7.73 | ||||||||
| 3 | 87.78 | 1.05 | 6.30 | 84.22 | 2.33 | 9.02 | |||||||||
| 10 | 94.78 | 0.54 | 8.55 | 84.10 | 3.56 | 10.28 | |||||||||
| OTA | 5 | 91.62 | 1.17 | 4.51 | 85.99 | 7.25 | 8.17 | ||||||||
| 10 | 91.97 | 0.19 | 4.67 | 81.94 | 2.33 | 9.49 | |||||||||
| 30 | 94.90 | 0.56 | 6.36 | 87.40 | 0.55 | 8.71 | |||||||||
| Method | Matrix | Mycotoxins | * R2 | LOQ | * RSD (%) | RE (%) | Ref. |
|---|---|---|---|---|---|---|---|
| QuEChERS-LC-MS/MS | High oil content (almonds, peanuts, walnuts, hazelnuts, pecan nuts, cashews) | AFG2 | >0.9942 | 1.25 | <20 | 73.66 | [36] |
| AFG1 | >0.9857 | 1.25 | <19 | 78.00 | |||
| AFB2 | >0.9938 | 1.25 | <14 | 80.00 | |||
| AFB1 | >0.9787 | 1.25 | <19 | 68.33 | |||
| OTA | >0.9939 | 5.00 | <17 | 76.00 | |||
| QuEChERS-LC-MS/MS | High oil content (sesame butter) | AFG2 | 0.9987 | 0.21 | <6 | 93.0 | [37] |
| AFG1 | 0.9979 | 0.21 | <3 | 95..0 | |||
| AFB2 | 0.9983 | 0.21 | <5 | 97.0 | |||
| AFB1 | 0.9991 | 0.21 | <5 | 99.9 | |||
| OTA | 0.9987 | 0.74 | - | - | |||
| QuEChERS-LC-MS/MS | High-sugar and high-water content (Grapes and Wines) | AFG2 | 0.9988 | 0.18 | <18 | 94.39 | [24] |
| AFG1 | 0.9988 | 0.75 | <16 | 87.95 | |||
| AFB2 | 0.9993 | 0.39 | <8 | 94.41 | |||
| AFB1 | 0.9990 | 0.75 | <11 | 100.29 | |||
| OTA | 0.9998 | 0.3 | <17 | 96.06 | |||
| QuEChERS-LC-MS/MS | Food containing complex components (Different species and medicinal herbs) | AFG2 | >0.9996 | 0.25 | <10 | 76.19 | [38] |
| AFG1 | >0.9947 | 1.00 | <9 | 82.58 | |||
| AFB2 | >0.9968 | 0.25 | <7 | 87.94 | |||
| AFB1 | >0.9933 | 1.00 | <10 | 84.39 | |||
| OTA | >0.9996 | 0.5 | <16 | 66.5 | |||
| QuEChERS-LC-MS/MS | Different food matrices | AFG2 | >0.9984 | 0.08-0.10 | <11 | 93.06 | Present work |
| AFG1 | >0.9989 | 0.13 | <11 | 99.11 | |||
| AFB2 | >0.9987 | 0.08-0.10 | <7 | 97.94 | |||
| AFB1 | >0.9989 | 0.08-0.13 | <6 | 99.04 | |||
| OTA | >0.9967 | 0.10-0.30 | <10 | 93.82 |
| Sample | NS | Concentration (µg/L for Liquid Samples & µg/kg for Non-Liquid Samples) | ||||
|---|---|---|---|---|---|---|
| AFB1 | AFB2 | AFG1 | AFG2 | OTA | ||
| Apple juice | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Grape juice | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Orange juice | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Pomegranate juice | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Raisin | 10 | 2.73, 5.67 | 0.84, 1.33 | 1.50, 2.50 | 1.47 | n.d. |
| Dried-fig | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Wheat flour | 10 | 1.50, 7.33, 10.12 | 0.45, 2.70 | 1.80, 2.61 | n.d. | 1.2 |
| Barley flour | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Non-roasted peanut | 10 | 5.36, 10.23 | 1.45, 2.22 | 2.00, 4.35 | 0.76, 0.82 | 1.20, 3.53 |
| Roasted pistachio | 10 | 5.30, 5.48, 7.48, 10.15 | 1.46, 1.60, 3.47 | 1.90, 2.1, 2.5, 3.31 | 0.81, 0.90 | n.d. |
| Chili | 10 | 4.90, 5.26, 8.70, 16.93 | 1.45, 4.69, 8.11 | 1.76, 1.89, 2.10, 6.96 | 0.71, 0.96 | n.d. |
| Mixed spice | 10 | 4.70, 7.41, 10.69, 14.36 | 1.52, 2.26, 3.43, 4.13 | 1.55, 1.79, 7.74 | n.d. | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsharif, A.M.A.; Choo, Y.-M.; Tan, G.-H. Detection of Five Mycotoxins in Different Food Matrices in the Malaysian Market by Using Validated Liquid Chromatography Electrospray Ionization Triple Quadrupole Mass Spectrometry. Toxins 2019, 11, 196. https://doi.org/10.3390/toxins11040196
Alsharif AMA, Choo Y-M, Tan G-H. Detection of Five Mycotoxins in Different Food Matrices in the Malaysian Market by Using Validated Liquid Chromatography Electrospray Ionization Triple Quadrupole Mass Spectrometry. Toxins. 2019; 11(4):196. https://doi.org/10.3390/toxins11040196
Chicago/Turabian StyleAlsharif, Ali Mohamed Ali, Yeun-Mun Choo, and Guan-Huat Tan. 2019. "Detection of Five Mycotoxins in Different Food Matrices in the Malaysian Market by Using Validated Liquid Chromatography Electrospray Ionization Triple Quadrupole Mass Spectrometry" Toxins 11, no. 4: 196. https://doi.org/10.3390/toxins11040196
APA StyleAlsharif, A. M. A., Choo, Y.-M., & Tan, G.-H. (2019). Detection of Five Mycotoxins in Different Food Matrices in the Malaysian Market by Using Validated Liquid Chromatography Electrospray Ionization Triple Quadrupole Mass Spectrometry. Toxins, 11(4), 196. https://doi.org/10.3390/toxins11040196