The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function



Abstract
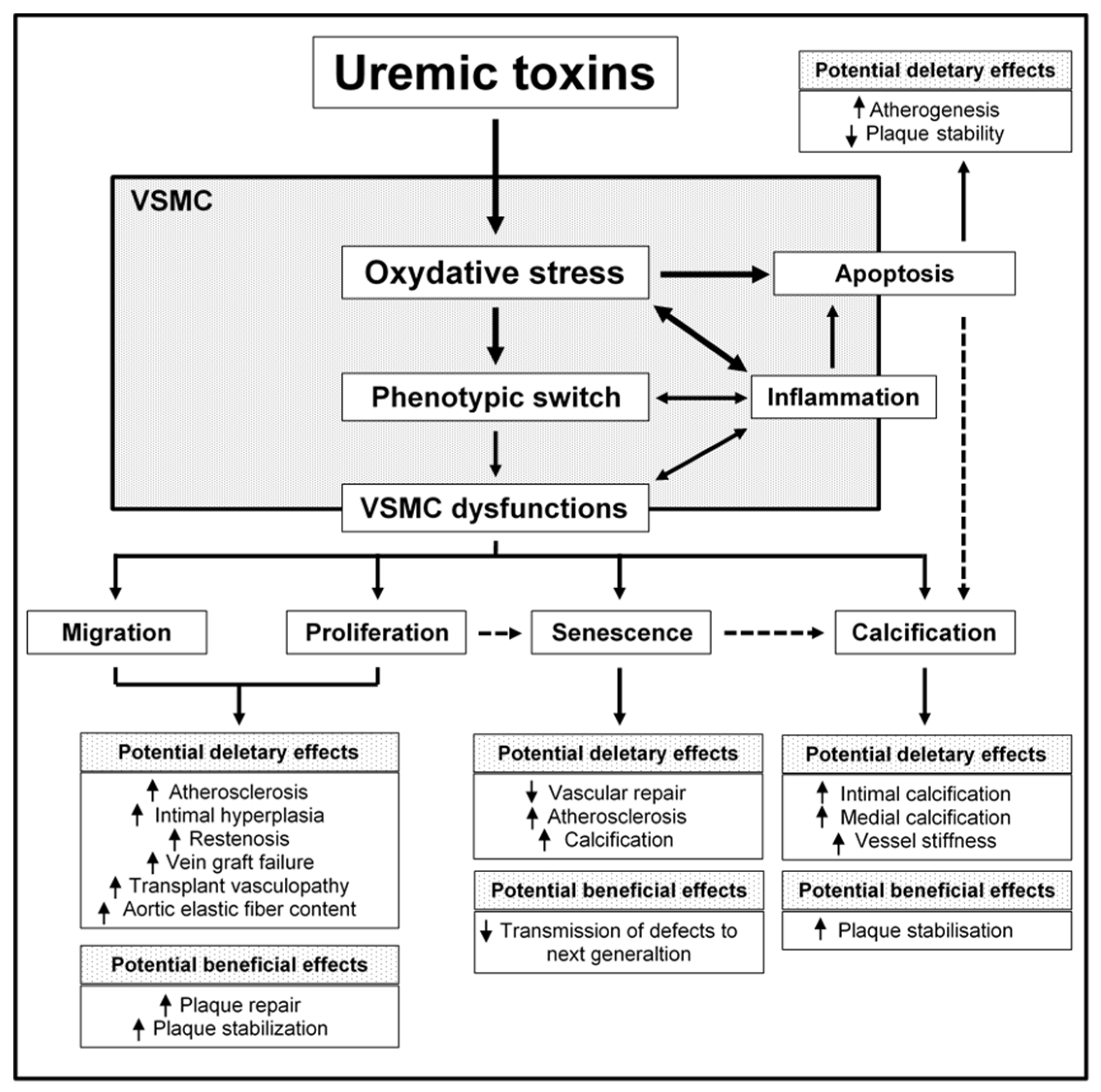
1. Introduction
2. The Impact of Uremic Toxins on VSMC Function
2.1. Proliferation
2.2. Migration
2.3. Cell Death
2.4. Calcification and Osteogenic Differentiation
2.5. Senescence
3. Conclusions
Funding
Conflicts of Interest
References
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Brunet, P.; Gondouin, B.; Duval-Sabatier, A.; Dou, L.; Cerini, C.; Dignat-George, F.; Jourde-Chiche, N.; Argiles, A.; Burtey, S. Does uremia cause vascular dysfunction? Kidney Blood Press Res. 2011, 34, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Lu, L.; Hua, Y.; Huang, K.; Wang, I.; Huang, L.; Fu, Q.; Chen, A.; Chan, P.; Fan, H.; et al. Vasculopathy in the setting of cardiorenal syndrome: Roles of protein-bound uremic toxins. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1–H13. [Google Scholar] [CrossRef] [PubMed]
- Monroy, M.A.; Fang, J.; Li, S.; Ferrer, L.; Birkenbach, M.P.; Lee, I.J.; Wang, H.; Yang, X.F.; Choi, E.T. Chronic kidney disease alters vascular smooth muscle cell phenotype. Front. Biosci. (Landmark Ed.) 2015, 20, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.; Aarup, A.; Albinsson, S.; Hartvigsen, K.; Sørensen, C.M.; Turczynska, K.; Nielsen, L.B.; Pedersen, T.X. Uremia modulates the phenotype of aortic smooth muscle cells. Atherosclerosis 2017, 257, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Cazaña-Pérez, V.; Cidad, P.; Donate-Correa, J.; Martín-Núñez, E.; López-López, J.R.; Pérez-García, M.T.; Giraldez, T.; Navarro-González, J.F.; Alvarez de la Rosa, D. Phenotypic Modulation of Cultured Primary Human Aortic Vascular Smooth Muscle Cells by Uremic Serum. Front. Physiol. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Marchais, S.J.; Safar, M.E.; Genest, A.F.; Guerin, A.P.; Metivier, F.; Chedid, K.; London, A.M. Aortic and large artery compliance in end-stage renal failure. Kidney Int. 1990, 37, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Niwa, T. Indoxyl sulfate promotes proliferation of human aortic smooth muscle cells by inducing oxidative stress. J. Ren. Nutr. 2009, 19, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Hirose, Y.; Nishijima, F.; Tsubakihara, Y.; Miyazaki, H. ROS and PDGF-beta [corrected] receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am. J. Physiol. Cell Physiol. 2009, 297, C389–C396. [Google Scholar] [CrossRef] [PubMed]
- Yisireyili, M.; Saito, S.; Abudureyimu, S.; Adelibieke, Y.; Ng, H.Y.; Nishijima, F.; Takeshita, K.; Murohara, T.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS ONE 2014, 9, e109268. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Bolati, W.; Lee, C.T.; Chien, Y.S.; Yisireyili, M.; Saito, S.; Pei, S.N.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Downregulates Mas Receptor via Aryl Hydrocarbon Receptor/Nuclear Factor-kappa B, and Induces Cell Proliferation and Tissue Factor Expression in Vascular Smooth Muscle Cells. Nephron 2016, 133, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Hsu, S.C.; Lee, H.S.; Lin, S.H.; Tsai, C.S.; Huang, S.M.; Shih, C.C.; Hsu, Y.J. Enhanced expression of glucose transporter-1 in vascular smooth muscle cells via the Akt/tuberous sclerosis complex subunit 2 (TSC2)/mammalian target of rapamycin (mTOR)/ribosomal S6 protein kinase (S6K) pathway in experimental renal failure. J. Vasc. Surg. 2013, 57, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Mozar, A.; Louvet, L.; Morlière, P.; Godin, C.; Boudot, C.; Kamel, S.; Drüeke, T.B.; Massy, Z.A. Uremic toxin indoxyl sulfate inhibits human vascular smooth muscle cell proliferation. Ther. Apher. Dial. 2011, 15, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol. Cell Physiol 2012, 303, C126–C134. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Chen, Y.; Zhu, Z.; Su, X.; Ni, J.; Du, R.; Zhang, R.; Jin, W. p-Cresyl sulfate promotes the formation of atherosclerotic lesions and induces plaque instability by targeting vascular smooth muscle cells. Front. Med. 2016, 10, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; van Ypersele de Strihou, C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997, 51, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, Z.; Gong, K.; Zhao, P.; Qin, J.; Liu, N. Inhibition of reactive oxygen species/extracellular signal-regulated kinases pathway by pioglitazone attenuates advanced glycation end products-induced proliferation of vascular smooth muscle cells in rats. Biol. Pharm. Bull. 2011, 34, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Yoon, Y.W.; Lee, B.K.; Kwon, H.M.; Hwang, K.C.; Kim, M.; Chang, W.; Hong, B.K.; Lee, Y.H.; Park, S.J.; et al. Potential role of HMG CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp. Mol. Med. 2009, 41, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Togo, M.; Hara, M.; Miyata, T.; Han, K.; Maekawa, H.; Ohno, M.; Hashimoto, Y.; Kurokawa, K.; Watanabe, T. Advanced glycation endproducts stimulate mitogen-activated protein kinase and proliferation in rabbit vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1997, 239, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Li, S.L.; Sahar, S.; Kim, Y.S.; Xu, Z.G.; Lanting, L.; Natarajan, R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J. Biol. Chem. 2006, 281, 13685–13693. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, M.S.; Fernández, J.M.; Cortizo, A.M.; McCarthy, A.D.; Schurman, L.; Sedlinsky, C. Advanced glycation end products and strontium ranelate promote osteogenic differentiation of vascular smooth muscle cells in vitro: Preventive role of vitamin D. Mol. Cell. Endocrinol. 2017, 450, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Yan, S.F.; Yan, S.D.; Belov, D.; Rong, L.L.; Sousa, M.; Andrassy, M.; Marso, S.P.; Duda, S.; Arnold, B.; et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Investig. 2003, 111, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, K.; Penn, M.S.; Marso, S.P.; Lauer, M.A.; Forudi, F.; Zhou, X.; Qu, W.; Lu, Y.; Stern, D.M.; et al. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation 2003, 107, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Eun, S.Y.; Ko, Y.S.; Park, S.W.; Chang, K.C.; Kim, H.J. IL-1β enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor-mediated RAGE expression and HMGB1 release. Vasc. Pharmacol. 2015, 72, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cheng, Y.; Simoncini, T.; Xu, S. 17β-Estradiol inhibits TNF-α-induced proliferation and migration of vascular smooth muscle cells via suppression of TRAIL. Gynecol. Endocrinol. 2016, 32, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, U.; Ikeda, M.; Oohara, T.; Kano, S.; Yaginuma, T. Mitogenic action of interleukin-1 alpha on vascular smooth muscle cells mediated by PDGF. Atherosclerosis 1990, 84, 183–188. [Google Scholar] [CrossRef]
- Selzman, C.H.; Shames, B.D.; Reznikov, L.L.; Miller, S.A.; Meng, X.; Barton, H.A.; Werman, A.; Harken, A.H.; Dinarello, C.A.; Banerjee, A. Liposomal delivery of purified inhibitory-kappaBalpha inhibits tumor necrosis factor-alpha-induced human vascular smooth muscle proliferation. Circ. Res. 1999, 84, 867–875. [Google Scholar] [CrossRef] [PubMed]
- OuYang, P.; Peng, L.S.; Yang, H.; Wu, W.Y.; Xu, A.L. Recombinant human interleukin-10 inhibits vascular smooth muscle cell proliferation induced by TNF-alpha. Sheng Li Xue Bao 2002, 54, 79–82. [Google Scholar] [PubMed]
- Tang, H.P.; Sun, L.X.; Han, W. Endothelial cells on the proliferation and expression of intercellular adhesion molecule 1 and interleukin 8 of vascular smooth muscle cells. Genet. Mol. Res. 2013, 12, 4363–4370. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A.; Hajjar, D.P.; Silverstein, R.L.; Nachman, R.L. Tumor necrosis factor-mediated release of platelet-derived growth factor from cultured endothelial cells. J. Exp. Med. 1987, 166, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Metzinger-Le Meuth, V.; Hénaut, L.; Djelouat, M.S.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: Evidence for the involvement of miR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef] [PubMed]
- Rahabi-Layachi, H.; Ourouda, R.; Boullier, A.; Massy, Z.A.; Amant, C. Distinct Effects of Inorganic Phosphate on Cell Cycle and Apoptosis in Human Vascular Smooth Muscle Cells. J. Cell. Physiol. 2015, 230, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Ishikawa, N.; Uchida, H.; Chasnoff, S.E.; Xie, X.; Mathew, S.; Hruska, K.A.; Choi, E.T. CKD accelerates development of neointimal hyperplasia in arteriovenous fistulas. J. Am. Soc. Nephrol. 2009, 20, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Sano, H.; Saishoji, T.; Ikeda, K.; Jinnouchi, Y.; Kanzaki, T.; Morisaki, N.; Rauvala, H.; Shichiri, M.; Horiuchi, S. The receptor for advanced glycation end products mediates the chemotaxis of rabbit smooth muscle cells. Diabetes 1997, 46, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Yang, R.; Zhang, Y.; Wu, P.; Wang, L.; Gao, Z.; Wang, J. Effect of crocetin on vascular smooth muscle cells migration induced by advanced glycosylation end products. Microvasc. Res. 2017, 112, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Guignabert, C.; de Jesus Perez, V.; Alastalo, T.P.; Powers, J.M.; Wang, L.; Lawrie, A.; Ambartsumian, N.; Schmidt, A.M.; Berryman, M.; et al. S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ. Res. 2009, 105, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.M.; Su, X.L.; Wang, Y.; Li, G.R.; Deng, X.L. KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation endproducts in cultured rat vascular smooth muscle cells. Lab. Investig. 2013, 93, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.X.; Rai, V.; Shen, X.; Rosario, R.; Lu, Y.; D’Agati, V.; Yan, S.F.; Friedman, R.A.; Nuglozeh, E.; Schmidt, A.M. Activation of the ROCK1 branch of the transforming growth factor-beta pathway contributes to RAGE-dependent acceleration of atherosclerosis in diabetic ApoE-null mice. Circ. Res. 2010, 106, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Choi, H.J.; Kim, C.H.; Jeong, H.S.; Ha, K.T. Lipocalin-2 elicited by advanced glycation end-products promotes the migration of vascular smooth muscle cells. Biochim. Biophys. Acta 2013, 1833, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yamagishi, S.; Hsu, C.C.; Yamamoto, H. Advanced glycation endproducts-receptor interactions stimulate the growth of human pancreatic cancer cells through the induction of platelet-derived growth factor-B. Biochem. Biophys. Res. Commun. 1996, 222, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wu, L.; Li, B.; Meng, X.; Dai, H.; Zheng, Y.; Fu, J. Cyanidin-3-O-glucoside Induces Apoptosis and Inhibits Migration of Tumor Necrosis Factor-α-Treated Rat Aortic Smooth Muscle Cells. Cardiovasc. Toxicol. 2016, 16, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Xu, W.; Guo, L.; Ning, W.; Zeng, X. Paeonol Inhibits the Proliferation, Invasion, and Inflammatory Reaction Induced by TNF-α in Vascular Smooth Muscle Cells. Cell Biochem. Biophys. 2015, 73, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Chang, Y.C.; Whang, K.; Kim, C.H.; Lee, I.S. Role of NAD(P)H:quinone oxidoreductase 1 on tumor necrosis factor-alpha-induced migration of human vascular smooth muscle cells. Cardiovasc. Res. 2007, 76, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Sun, Q.; Huang, L.; Meng, G.; Wang, H.; Jing, X.; Zhang, W. Tetrahydroxystilbene glucoside inhibits TNF-α-induced migration of vascular smooth muscle cells via suppression of vimentin. Can. J. Physiol. Pharmacol. 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Goetze, S.; Xi, X.P.; Kawano, Y.; Kawano, H.; Fleck, E.; Hsueh, W.A.; Law, R.E. TNF-alpha-induced migration of vascular smooth muscle cells is MAPK dependent. Hypertension 1999, 33 Pt 2, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Ichiki, T.; Fukuyama, K.; Iino, N.; Masuda, S.; Egashira, K.; Takeshita, A. cAMP-response element-binding protein mediates tumor necrosis factor-alpha-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.H.; Castresana, M.R.; Webb, J.G.; Wang, Z. Cyclic AMP inhibits production of interleukin-6 and migration in human vascular smooth muscle cells. J. Surg. Res. 2003, 109, 57–61. [Google Scholar] [CrossRef]
- Wang, Z.; Newman, W.H. Smooth muscle cell migration stimulated by interleukin 6 is associated with cytoskeletal reorganization. J. Surg. Res. 2003, 111, 261–266. [Google Scholar] [CrossRef]
- Wang, Z.; Kong, L.; Kang, J.; Vaughn, D.M.; Bush, G.D.; Walding, A.L.; Grigorian, A.A.; Robinson, J.S.; Nakayama, D.K. Interleukin-lβ induces migration of rat arterial smooth muscle cells through a mechanism involving increased matrix metalloproteinase-2 activity. J. Surg. Res. 2011, 169, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ. Res. 2008, 102, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell. 2011, 21, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Nakano, T.; Akishita, M.; Ouchi, Y. Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur. J. Pharmacol. 2007, 556, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Trécherel, E.; Godin, C.; Louandre, C.; Benchitrit, J.; Poirot, S.; Mazière, J.C.; Massy, Z.A.; Galmiche, A. Upregulation of BAD, a pro-apoptotic protein of the BCL2 family, in vascular smooth muscle cells exposed to uremic conditions. Biochem. Biophys. Res. Commun. 2012, 417, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Bowen-Pope, D.F.; Schaub, F.J. Apoptosis of smooth muscle cells is not silent: Fas/FADD initiates a program of inflammatory gene expression. Trends Cardiovasc. Med. 2001, 11, 42–45. [Google Scholar] [CrossRef]
- Schaub, F.J.; Han, D.K.; Liles, W.C.; Adams, L.D.; Coats, S.A.; Ramachandran, R.K.; Seifert, R.A.; Schwartz, S.M.; Bowen-Pope, D.F. Fas/FADD-mediated activation of a specific program of inflammatory gene expression in vascular smooth muscle cells. Nat. Med. 2000, 6, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Chang, J.S.; Baek, J.A.; Chung, M.Y.; Lee, H.C.; Rhim, B.Y.; Sok, D.E.; Rho, M.C.; Kim, Y.K.; Kim, K. TNF-alpha activates death pathway in human aorta smooth muscle cell in the presence of 7-ketocholesterol. Biochem. Biophys. Res. Commun. 2005, 333, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Obara, H.; Takayanagi, A.; Hirahashi, J.; Tanaka, K.; Wakabayashi, G.; Matsumoto, K.; Shimazu, M.; Shimizu, N.; Kitajima, M. Overexpression of truncated IkappaBalpha induces TNF-alpha-dependent apoptosis in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Pillai, S.; Lawrence, N.; Sebti, S.; Chellappan, S.P. TNF-α-mediated proliferation of vascular smooth muscle cells involves Raf-1-mediated inactivation of Rb and transcription of E2F1-regulated genes. Cell Cycle 2012, 11, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Tang, V.; Dhirapong, A.; Yabes, A.P.; Weiss, R.H. TNF-alpha-mediated apoptosis in vascular smooth muscle cells requires p73. Am. J. Physiol. Cell Physiol. 2005, 289, C199–C206. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Fang, J. TNF-α regulates apoptosis of human vascular smooth muscle cells through gap junctions. Mol. Med. Rep. 2017, 15, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Wu, Q.; Muszynski, M.; Hansson, G.K.; Libby, P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin-1 beta. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Kusano, E.; Akimoto, T.; Takeda, S.; Sasaki, N.; Umino, T.; Iimura, O.; Ando, Y.; Asano, Y. Cilostazol enhances IL-1beta-induced NO production and apoptosis in rat vascular smooth muscle via PKA-dependent pathway. Cell. Signal. 2002, 14, 625–632. [Google Scholar] [CrossRef]
- Fukuo, K.; Hata, S.; Suhara, T.; Nakahashi, T.; Shinto, Y.; Tsujimoto, Y.; Morimoto, S.; Ogihara, T. Nitric oxide induces upregulation of Fas and apoptosis in vascular smooth muscle. Hypertension 1996, 27 Pt 2, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Rosner, D.; Stoneman, V.; Littlewood, T.; McCarthy, N.; Figg, N.; Wang, Y.; Tellides, G.; Bennett, M. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am. J. Pathol. 2006, 168, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shu, C.; Su, J.; Li, X. A crosstalk triggered by hypoxia and maintained by MCP-1/miR-98/IL-6/p38 regulatory loop between human aortic smooth muscle cells and macrophages leads to aortic smooth muscle cells apoptosis via Stat1 activation. Int. J. Clin. Exp. Pathol. 2015, 8, 2670–2679. [Google Scholar] [PubMed]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transplant. 2017. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Yano, S.; Tanaka, S.; Sheikh, A.M.; Nagai, A.; Sugimoto, T. Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Maltais, J.S.; Simard, E.; Froehlich, U.; Denault, J.B.; Gendron, L.; Grandbois, M. iRAGE as a novel carboxymethylated peptide that prevents advanced glycation end product-induced apoptosis and endoplasmic reticulum stress in vascular smooth muscle cells. Pharmacol. Res. 2016, 104, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]
- West, S.L.; Swan, V.J.; Jamal, S.A. Effects of calcium on cardiovascular events in patients with kidney disease and in a healthy population. Clin. J. Am. Soc. Nephrol. 2010, 5 (Suppl. S1), S41–S47. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Klassen, P.S.; Lazarus, J.M.; Ofsthun, N.; Lowrie, E.G.; Chertow, G.M. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 2004, 15, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamashita, M.; Hayashi, M. Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J. Biol. Chem. 2012, 287, 25706–25714. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Daoudi, M.; Rosa, M.; Vinod, M.; Louvet, L.; Copin, C.; Fanchon, M.; Vanhoutte, J.; Derudas, B.; Belloy, L.; et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ. Res. 2017, 121, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem. Biophys. Res. Commun. 2010, 391, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z.; Shao, J.S.; Lai, C.F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.L.; Towler, D.A. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr−/− mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Xu, M.J.; Zhao, M.M.; Dai, X.Y.; Kong, W.; Wilson, G.M.; Guan, Y.; Wang, C.Y.; Wang, X. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. 2012, 82, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Miyazaki-Anzai, S.; Levi, M.; Ting, T.C.; Miyazaki, M. PERK-eIF2α-ATF4-CHOP signaling contributes to TNFα-induced vascular calcification. J. Am. Heart Assoc. 2013, 2, e000238. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Quiñones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Son, B.K.; Akishita, M.; Iijima, K.; Kozaki, K.; Maemura, K.; Eto, M.; Ouchi, Y. Adiponectin antagonizes stimulatory effect of tumor necrosis factor-alpha on vascular smooth muscle cell calcification: Regulation of growth arrest-specific gene 6-mediated survival pathway by adenosine 5′-monophosphate-activated protein kinase. Endocrinology 2008, 149, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Massy, Z.A. New insights into the key role of interleukin 6 in vascular calcification of chronic kidney disease. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Watson, A.D.; Ji, S.; Boström, K.I. Heat shock protein 70 enhances vascular bone morphogenetic protein-4 signaling by binding matrix Gla protein. Circ. Res. 2009, 105, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chang, Q.; Xin, M.; Wang, Q.; Li, H.; Qian, J. Endogenous bone morphogenetic protein 2 plays a role in vascular smooth muscle cell calcification induced by interleukin 6 in vitro. Int. J. Immunopathol. Pharmacol. 2017, 30, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Helas, S.; Goettsch, C.; Schoppet, M.; Zeitz, U.; Hempel, U.; Morawietz, H.; Kostenuik, P.J.; Erben, R.G.; Hofbauer, L.C. Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am. J. Pathol. 2009, 175, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Callegari, A.; Coons, M.L.; Ricks, J.L.; Rosenfeld, M.E.; Scatena, M. Increased Calcification in Osteoprotegerin-Deficient Smooth Muscle Cells: Dependence on Receptor Activator of NF-κB Ligand and Interleukin 6. J. Vasc. Res. 2014, 51, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Taki, K.; Takayama, F.; Tsuruta, Y.; Niwa, T. Oxidative stress, advanced glycation end product, and coronary artery calcification in hemodialysis patients. Kidney Int. 2006, 70, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, W.M.; Zhang, X.L.; He, H.Q.; Sun, X.L.; Zeng, H.; Xu, X.F.; Huang, L.; Zhu, Z.; Zhang, L.; et al. AGE/RAGE promotes thecalcification of human aortic smooth muscle cells via the Wnt/β-catenin axis. Am. J. Transl. Res. 2016, 8, 4644–4656. [Google Scholar] [PubMed]
- He, H.Q.; Liu, Y.; Zeng, H.; Sun, X.L.; Zhang, L.; Zhang, X.L.; Liao, W.J.; Zhou, X.Y.; He, Y.Z. Advanced glycation endproducts regulate smooth muscle cells calcification in cultured HSMCs. Int. J. Clin. Exp. Pathol. 2015, 8, 12260–12267. [Google Scholar] [PubMed]
- Suga, T.; Iso, T.; Shimizu, T.; Tanaka, T.; Yamagishi, S.; Takeuchi, M.; Imaizumi, T.; Kurabayashi, M. Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J. Atheroscler. Thromb. 2011, 18, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Gawdzik, J.; Mathew, L.; Kim, G.; Puri, T.S.; Hofmann Bowman, M.A. Vascular remodeling and arterial calcification are directly mediated by S100A12 (EN-RAGE) in chronic kidney disease. Am. J. Nephrol. 2011, 33, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Hofmann Bowman, M.A.; Gawdzik, J.; Bukhari, U.; Husain, A.N.; Toth, P.T.; Kim, G.; Earley, J.; McNally, E.M. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, M.R.; Bouvet, C.; Bouchard, S.; Moreau, S.; Leblond, J.; Deblois, D.; Moreau, P. Reduction of advanced-glycation end products levels and inhibition of RAGE signaling decreases rat vascular calcification induced by diabetes. PLoS ONE 2014, 9, e85922. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Han, X.; Wang, L.; Diao, Z.; Liu, W. Indoxyl sulfate promotes vascular smooth muscle cell calcification via the JNK/Pit-1 pathway. Ren. Fail. 2016, 38, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; Liu, S.; Tang, W.; Zhou, J.; Wang, Y.; Yao, X.; Quarles, L.D. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J. Am. Soc. Nephrol. 2007, 18, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Mackenzie, N.C.; Millan, J.L.; Farquharson, C.; MacRae, V.E. A protective role for FGF-23 in local defence against disrupted arterial wall integrity? Mol. Cell. Endocrinol. 2013, 372, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, R.; Kawakami-Mori, F.; Mu, S.; Hirohama, D.; Majtan, B.; Shimizu, Y.; Yatomi, Y.; Fukumoto, S.; Fujita, T.; Shimosawa, T. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 2014, 85, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [PubMed]
- Mori, D.; Matsui, I.; Shimomura, A.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Oka, T.; Kubota, K.; Yonemoto, S.; et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Pietrement, C.; Touré, F. Reconsidering the Lack of Urea Toxicity in Dialysis Patients. Semin. Dial. 2016, 29, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.; Barker, S.; Kurz, D.J.; Erusalimsky, J.D. Cellular senescence after single and repeated balloon catheter denudations of rabbit carotid arteries. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Takemura, A.; Iijima, K.; Ota, H.; Son, B.K.; Ito, Y.; Ogawa, S.; Eto, M.; Akishita, M.; Ouchi, Y. Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Troyano, N.; Nogal, M.D.; Mora, I.; Diaz-Naves, M.; Lopez-Carrillo, N.; Sosa, P.; Rodriguez-Puyol, D.; Olmos, G.; Ruiz-Torres, M.P. Hyperphosphatemia induces cellular senescence in human aorta smooth muscle cells through integrin linked kinase (ILK) up-regulation. Mech. Ageing Dev. 2015, 152, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Sarraj, B.; Ohnishi, M.; Densmore, M.J.; Taguchi, T.; Goetz, R.; Mohammadi, M.; Lanske, B.; Razzaque, M.S. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)—Mediated regulation of systemic phosphate homeostasis. FASEB J. 2009, 23, 433–441. [Google Scholar] [CrossRef] [PubMed]
- John, G.B.; Cheng, C.Y.; Kuro-o, M. Role of Klotho in aging, phosphate metabolism, and CKD. Am. J. Kidney Dis. 2011, 58, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Misao, R.; Hori, M.; Ichigo, S.; Fujimoto, J.; Tamaya, T. Corticosteroid-binding globulin mRNA levels in human uterine endometrium. Steroids 1994, 59, 603–607. [Google Scholar] [CrossRef]
- Andreassi, M.G. DNA damage, vascular senescence and atherosclerosis. J. Mol. Med. 2008, 86, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Giles, P.J.; Sheerin, A.N.; Smith, S.K.; Lawton, J.J.; Ostler, E.L.; Rhys-Williams, W.; Kipling, D.; Faragher, R.G. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp. Gerontol. 2009, 44, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kawagoe, Y.; Matsuda, T.; Ueda, Y.; Shimada, N.; Ebihara, I.; Koide, H. Oral ADSORBENT AST-120 decreases carotid intima-media thickness and arterial stiffness in patients with chronic renal failure. Kidney Blood Press. Res. 2004, 27, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Fukagawa, M.; Nishi, S. Effects of AST-120 on left ventricular mass in predialysis patients. Am. J. Nephrol. 2011, 33, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Kitamura, K.; Kono, K.; Nakai, K.; Fujii, H.; Nishi, S. Association between AST-120 and abdominal aortic calcification in predialysis patients with chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, M.; Mizobuchi, M.; Finch, J.L.; Nakamura, H.; Martin, D.R.; Slatopolsky, E. Blockage of the renin-angiotensin system attenuates mortality but not vascular calcification in uremic rats: Sevelamer carbonate prevents vascular calcification. Am. J. Nephrol. 2009, 29, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, I.G.; Joki, N.; Nguyen-Khoa, T.; Guerrera, I.C.; Maizel, J.; Benchitrit, J.; Machado dos Reis, L.; Edelman, A.; Lacour, B.; Jorgetti, V.; et al. Lanthanum carbonate, like sevelamer-HCl, retards the progression of vascular calcification and atherosclerosis in uremic apolipoprotein E-deficient mice. Nephrol. Dial. Transplant. 2012, 27, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Phan, O.; Ivanovski, O.; Nguyen-Khoa, T.; Mothu, N.; Angulo, J.; Westenfeld, R.; Ketteler, M.; Meert, N.; Maizel, J.; Nikolov, I.G.; et al. Sevelamer prevents uremia-enhanced atherosclerosis progression in apolipoprotein E-deficient mice. Circulation 2005, 112, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Jun, M.; Venkataraman, V.; Razavian, M.; Cooper, B.; Zoungas, S.; Ninomiya, T.; Webster, A.C.; Perkovic, V. Antioxidants for chronic kidney disease. Cochrane Database Syst. Rev. 2012, 10, CD008176. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [PubMed]
- Boaz, M.; Smetana, S.; Weinstein, T.; Matas, Z.; Gafter, U.; Iaina, A.; Knecht, A.; Weissgarten, Y.; Brunner, D.; Fainaru, M.; et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet 2000, 356, 1213–1218. [Google Scholar] [CrossRef]
- Tepel, M.; van der Giet, M.; Statz, M.; Jankowski, J.; Zidek, W. The antioxidant acetylcysteine reduces cardiovascular events in patients with end-stage renal failure: A randomized, controlled trial. Circulation 2003, 107, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Kneis, C.; Beck, W.; Boenisch, O.; Klefisch, F.; Deppisch, R.; Zickler, D.; Schindler, R. Elimination of middle-sized uremic solutes with high-flux and high-cut-off membranes: A randomized in vivo study. Blood Purif. 2013, 36, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; Capelli, I.; Angelini, M.L.; Valentini, C.; Baraldi, O.; Scolari, M.P.; Stefoni, S. Importance of vascular calcification in kidney transplant recipients. Am. J. Nephrol. 2014, 39, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Sidibé, A.; Fortier, C.; Desjardins, M.P.; Zomahoun, H.T.V.; Boutin, A.; Mac-Way, F.; De Serres, S.; Agharazii, M. Reduction of Arterial Stiffness After Kidney Transplantation: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Vipattawat, K.; Kitiyakara, C.; Phakdeekitcharoen, B.; Kantachuvesiri, S.; Sumethkul, V.; Jirasiritham, S.; Stitchantrakul, W.; Disthabanchong, S. Vascular calcification in long-term kidney transplantation. Nephrology (Carlton) 2014, 19, 251–256. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Uremic Toxins | Proliferation | Migration | Apoptosis | Calcification | Senescence | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Effect | Mechanisms of Action | Effect | Mechanisms of Action | Effect | Mechanisms of Action | Effect | Mechanisms of Action | Effect | Mechanisms of Action | |
| phosphate | − | G1/S Blocade ↓Cyclin E, CDK2 ↓Phospho pRb ↑p15, p21, p27 | + | ↑miR223 ↓miR143, miR145 ↑KLF4, KLF5 ↑versican ↑PDGFR | + | ↑Caspase 3 ↑BAD ↑FasL ↑TNF-R1 ↓Gas6 | + | ↑Osteogenic transition ↑MMP9 ↑Apoptotis/senescence ↑Procalcific MVs ↓miR143/miR145 ↑KLF4/↑miR223 | + | ↑ILK ↑ROS ↑p53, p16, p21 ↑G1/S Blocade ↓Phospho Rb ↑SAβG |
| IS | Acute + | ↑ROS ↑PDGFB-R ↑Cyclin D1, P21 ↓p53 ↑GLUT1 | + | ↑ROS ↑PDGFB-R | ND | + | ↑Osteogenic transition ↑Pit-1 ↓klotho | + | ↑ROS ↑p21, p53 ↑Prelamin A ↓FACE1 ↑SAβG | |
| Chronic − | ↑ROS ↑P21, P27 | |||||||||
| PCS | To be clarified | + | ↑MMP2/9 ↓TIMP1/2 | + | ↑Bax ↓Bcl2 | ND | ND | |||
| AGEs | + | ↑ROS ↑Inflammation | + | ↑ROS ↑MMP2/9 ↑TGF-β ↑Lipocalin 2 ↑PDGF ↑Inflammation | + | ↑ER stress ↑BAD ↑Caspase 9 | + | ↑ROS ↑Osteogenic transition | ND | |
| TNF-α | + | ↑ROS ↑TRAIL ↑IL-6 ↑PDGFβ | + | ↑MMP2/9 ↑TRAIL ↑NQO1 ↑IL-6 | + | ↑Caspase 3/8 ↑p73β ↓Cx43 ↑Fas | + | ↑Osteogenic transition ↓ANKH = PPi↓ ↑Pit-1 ↑ER stress ↓Gas6 = apoptosis↑ ↓Klotho ↑IL-6 | ND | |
| IL-6 | + | Mediates TNF-α effects | + | ↑Actin polymerization ↑Phospho FAK/paxillin Mediates TNF-α effects | + | ↑Stat1 Mediates TNF-α effects | + | ↑HSP70 ↑RANKL Mediates TNF-α effects | ND | |
| IL-1β | + | ↑PDGFβ ↑P2Y2R ↑RAGE ↑HMGB1 | + | ↑MMP2 ↑P2Y2R | + | ↑Fas ↑NO | + | Induction of TNAP | ND | |
| FGF-23 | ND | No influence | ND | To be clarified | ND | |||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hénaut, L.; Mary, A.; Chillon, J.-M.; Kamel, S.; Massy, Z.A. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. https://doi.org/10.3390/toxins10060218
Hénaut L, Mary A, Chillon J-M, Kamel S, Massy ZA. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins. 2018; 10(6):218. https://doi.org/10.3390/toxins10060218
Chicago/Turabian StyleHénaut, Lucie, Aurélien Mary, Jean-Marc Chillon, Saïd Kamel, and Ziad A. Massy. 2018. "The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function" Toxins 10, no. 6: 218. https://doi.org/10.3390/toxins10060218
APA StyleHénaut, L., Mary, A., Chillon, J.-M., Kamel, S., & Massy, Z. A. (2018). The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins, 10(6), 218. https://doi.org/10.3390/toxins10060218