Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
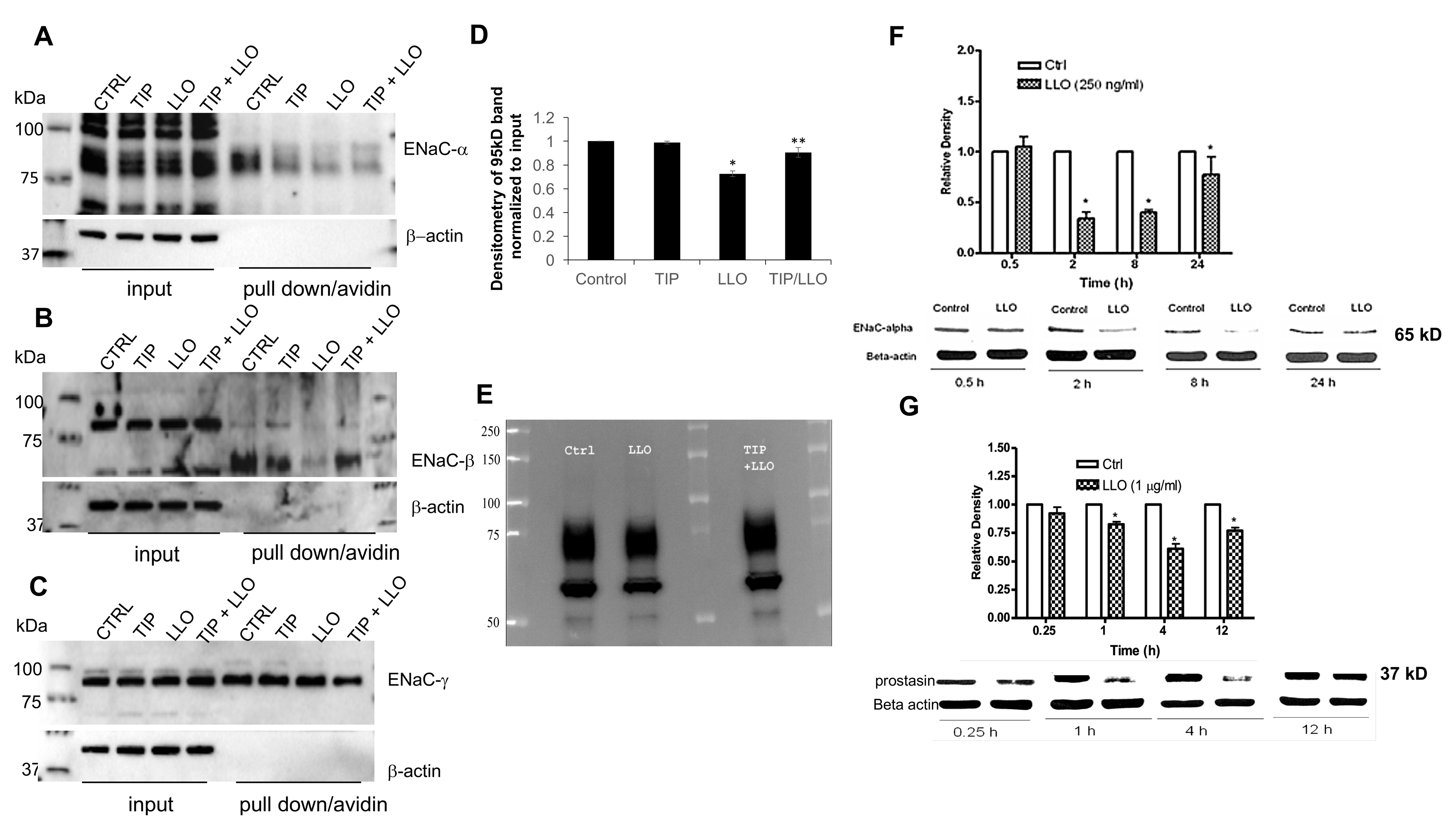
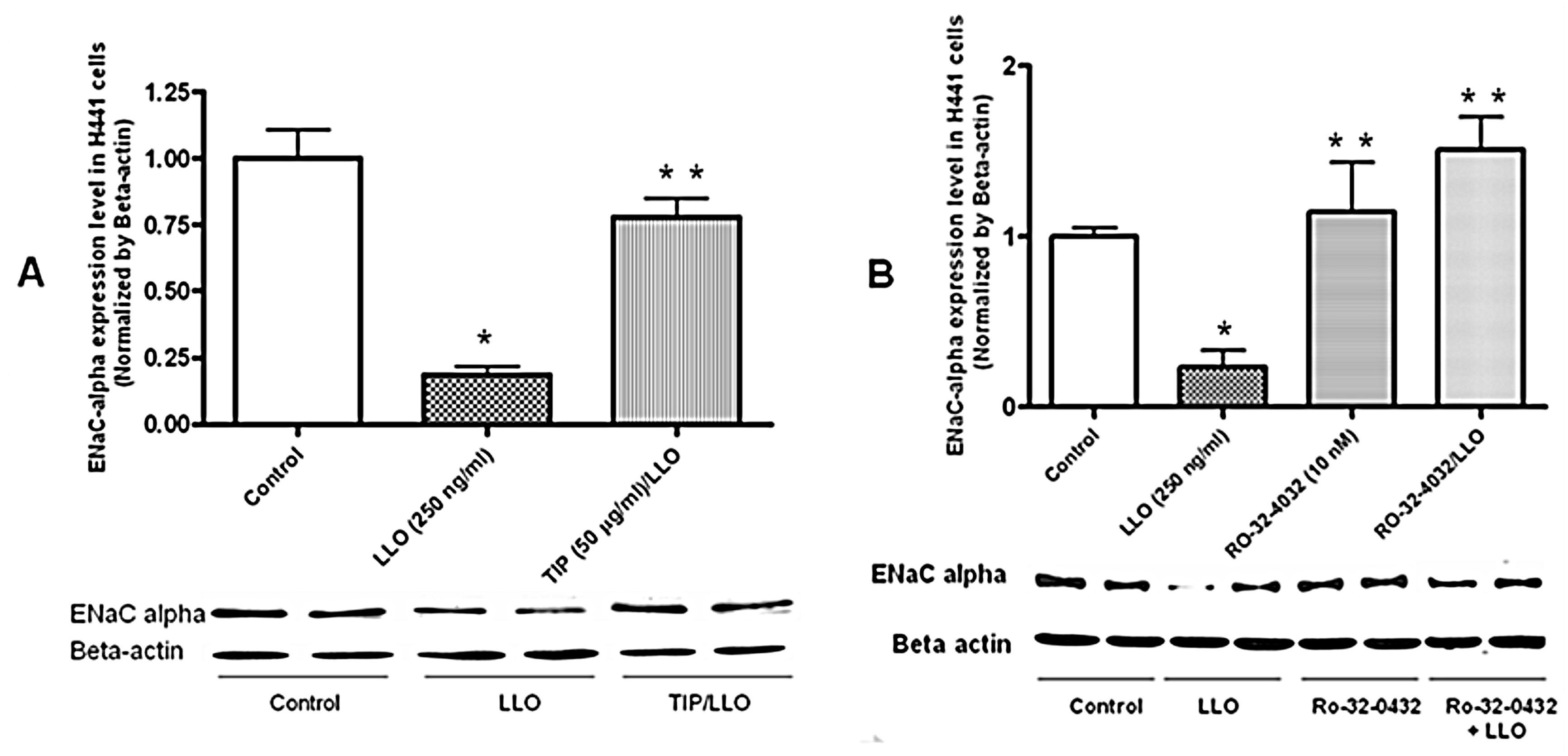
2.1. LLO Decreases ENaC-α Subunit Expression Levels in H441 Cells in a Time-Dependent Manner
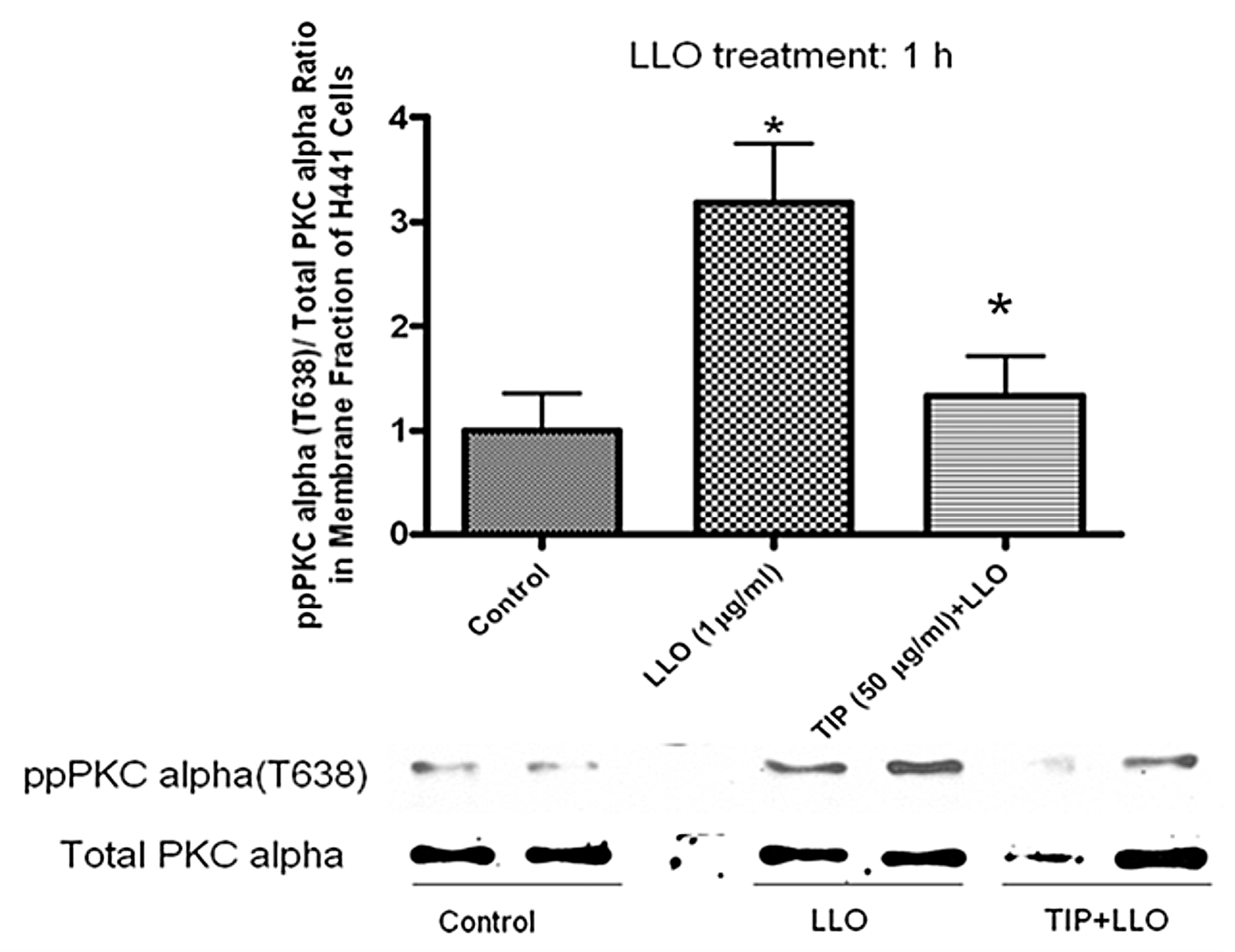
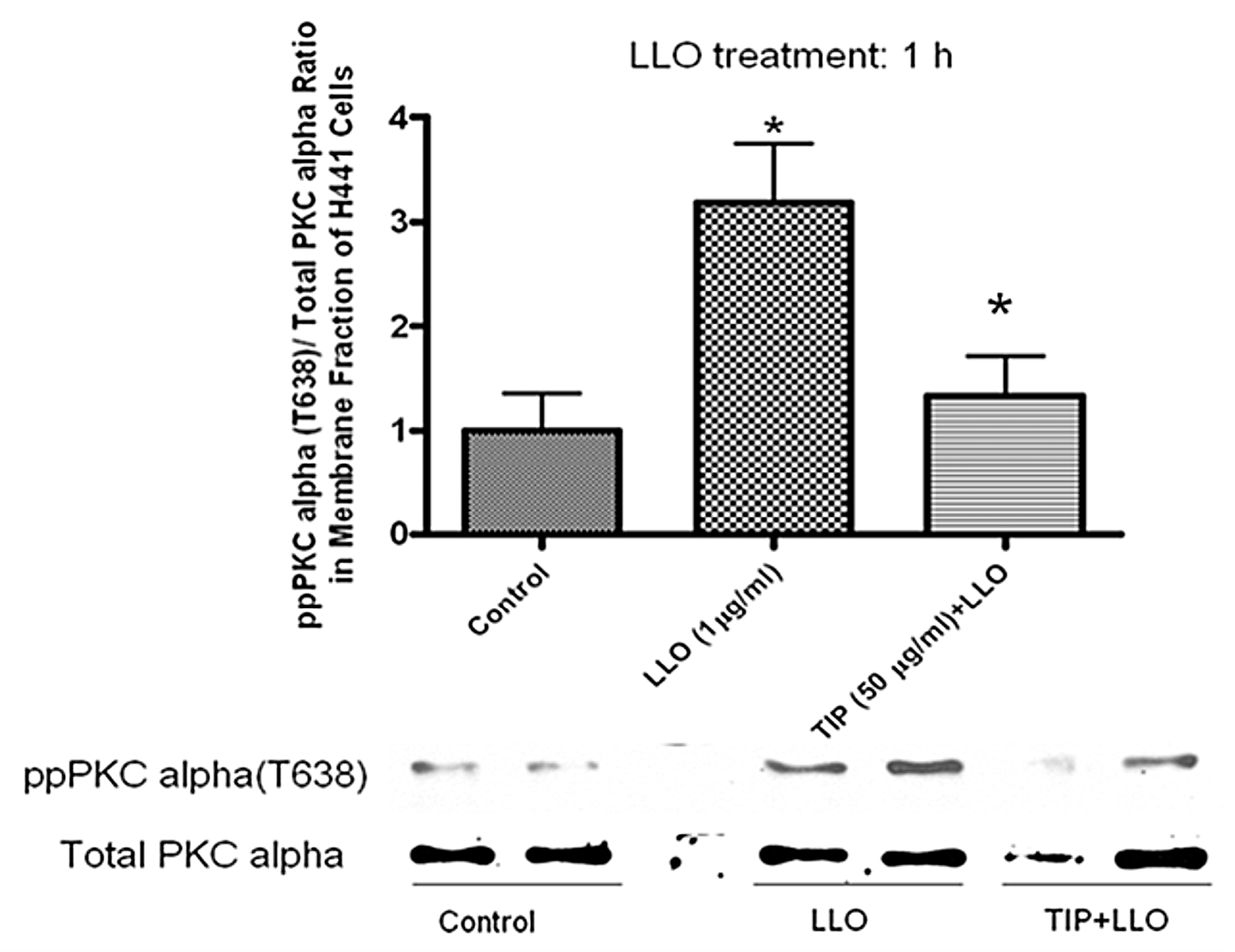
2.2. LLO-Induces PKC-α Activation in H441 Cells
2.3. The Protein Kinase C-α Inhibitor Ro-32-0432 Significantly Prevents LLO-Mediated Reduction of ENaC-α Expression in H441 Cells
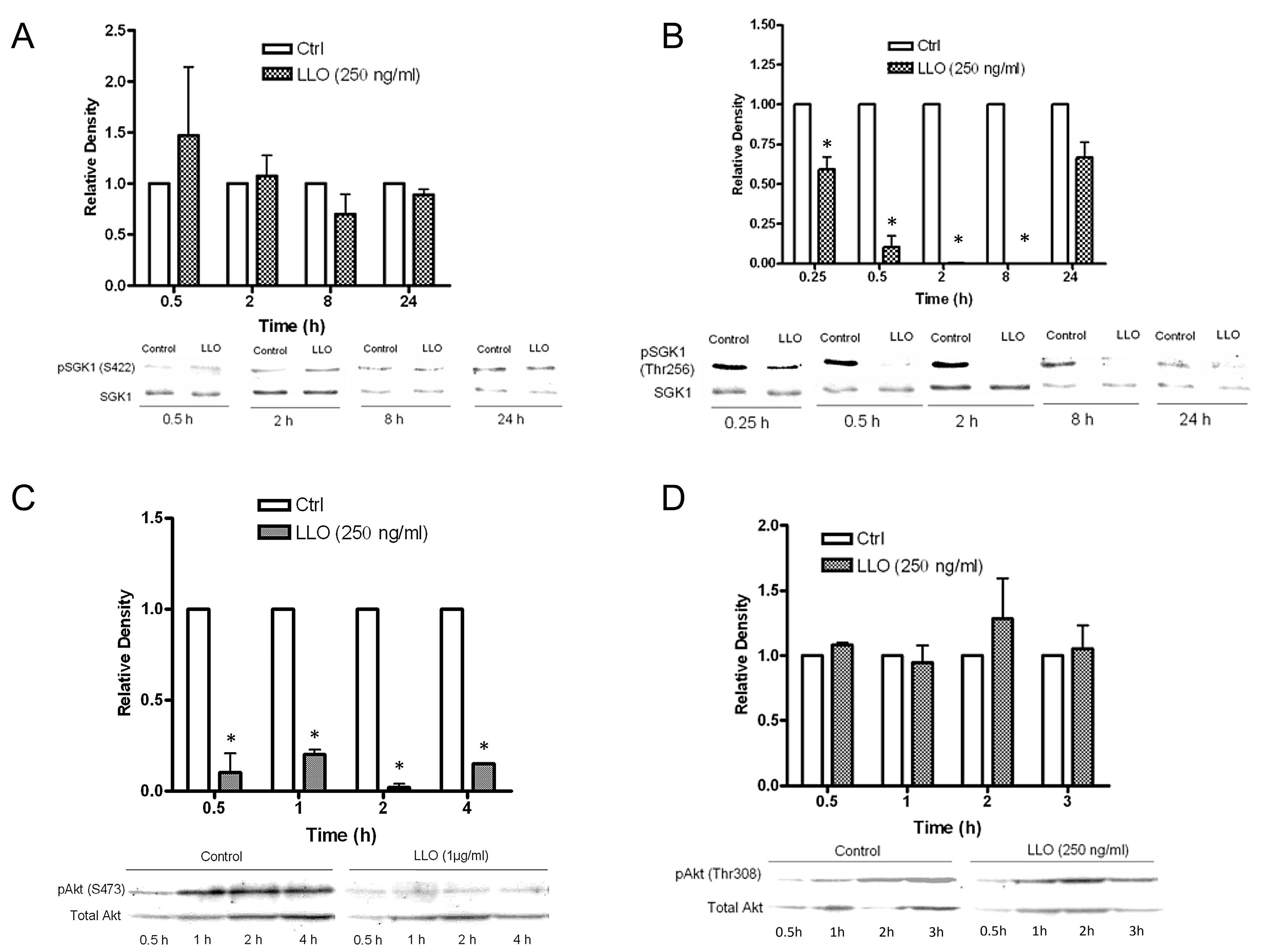
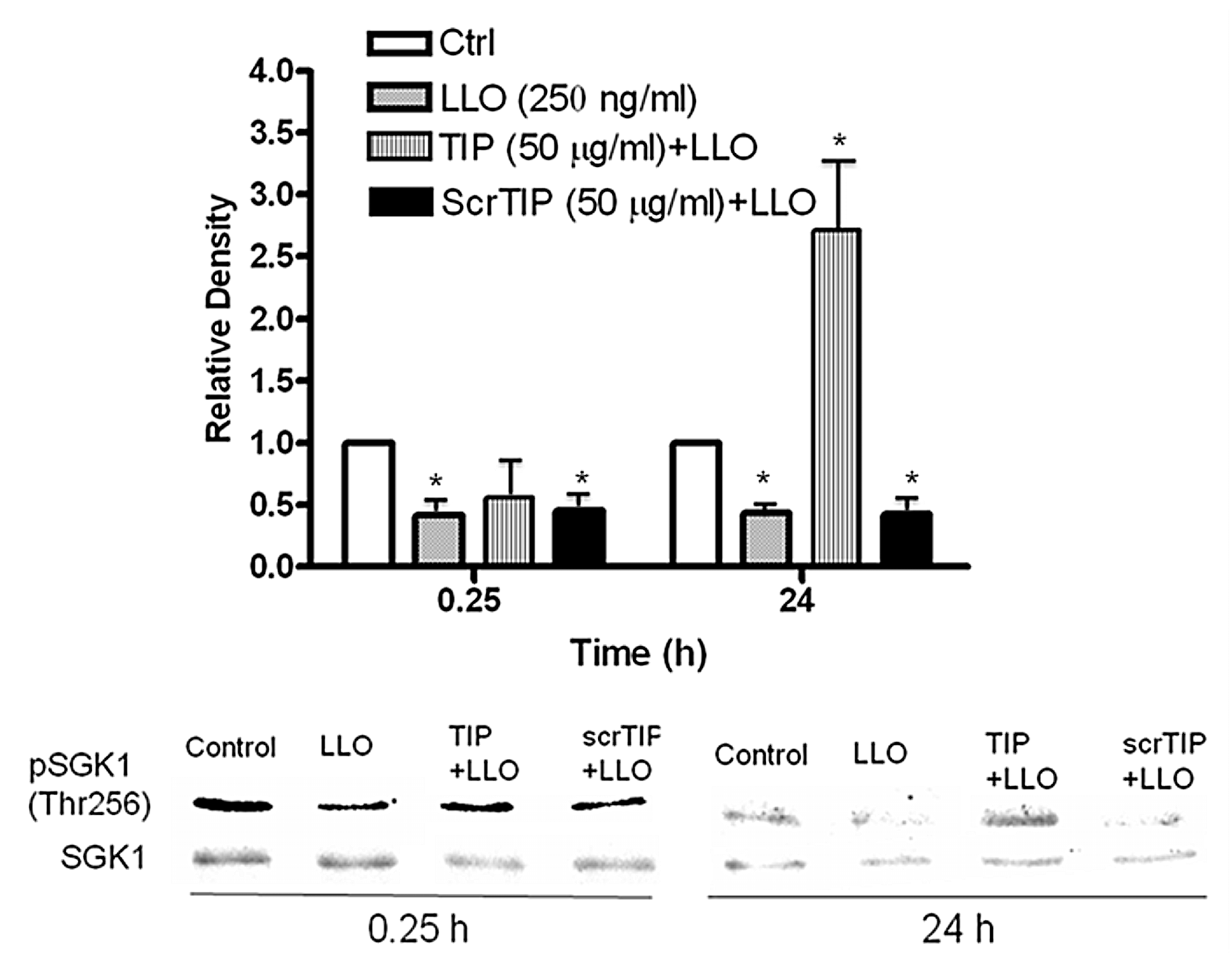
2.4. LLO Reduces Sgk-1 Phosphorylation at Residue T256 and Akt-1 Phosphorylation at Residue S473
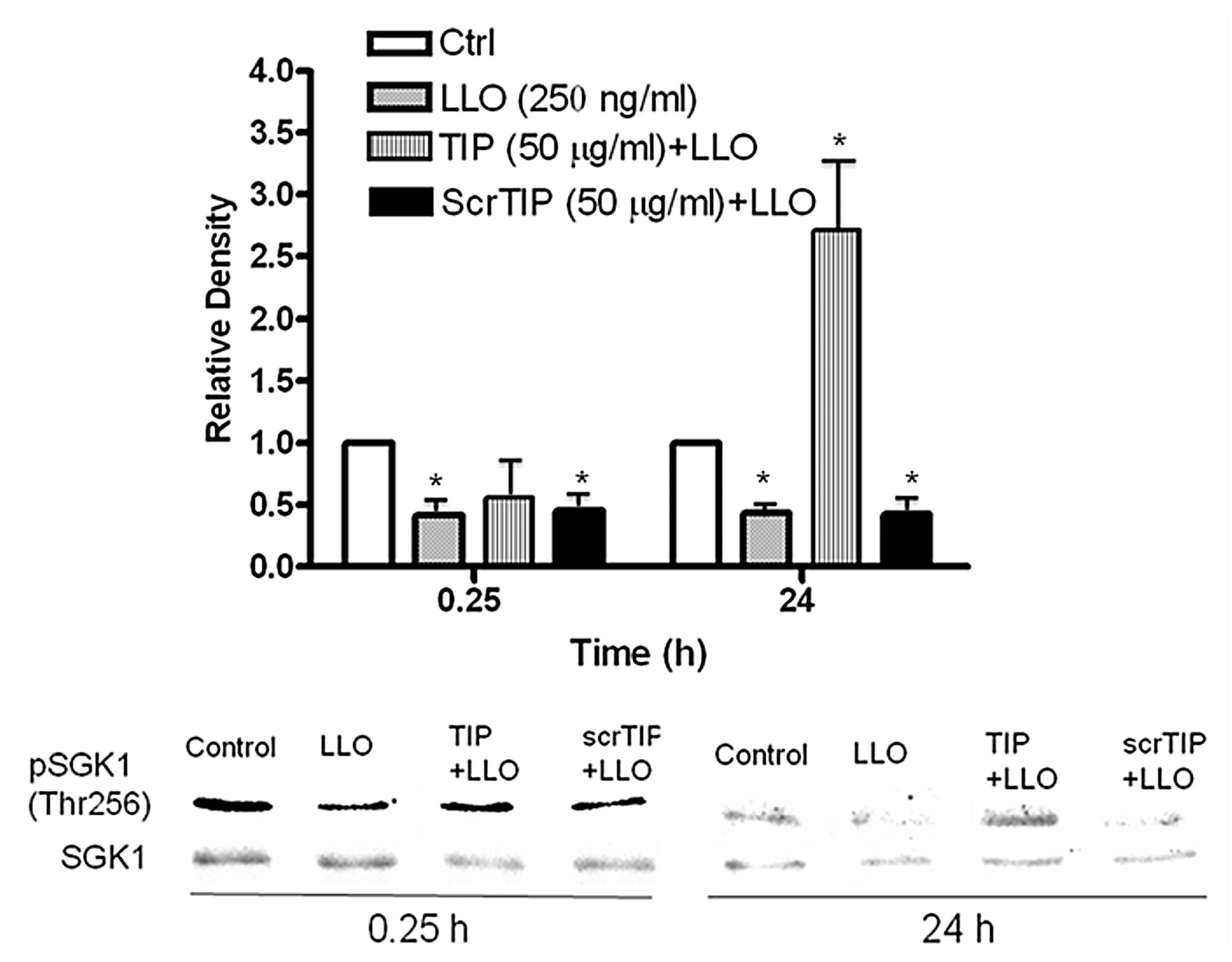
2.5. The TNF-Derived TIP Peptide Significantly Restores pT256Sgk-1 Expression Level at 24 h in LLO-Treated H441 Cells
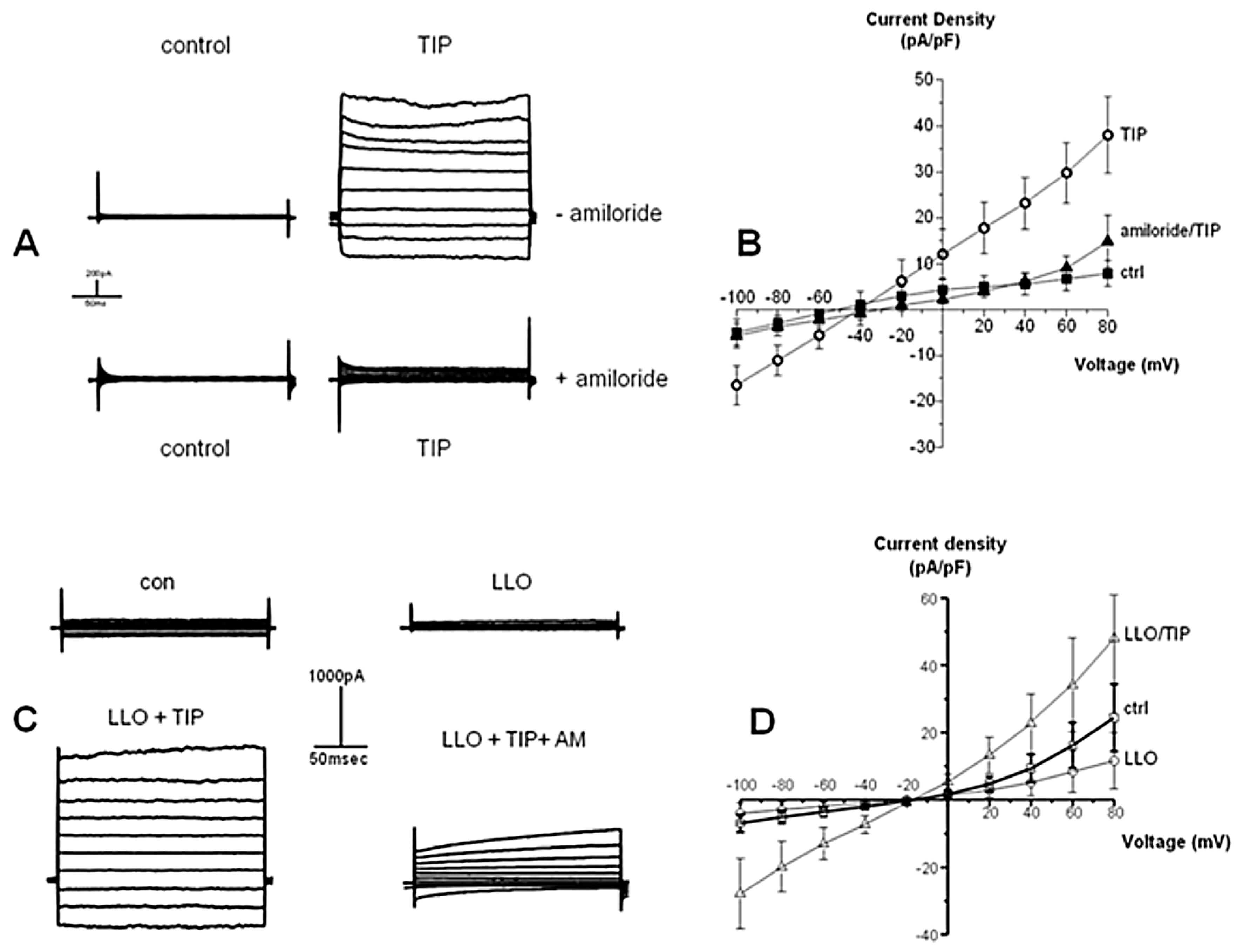
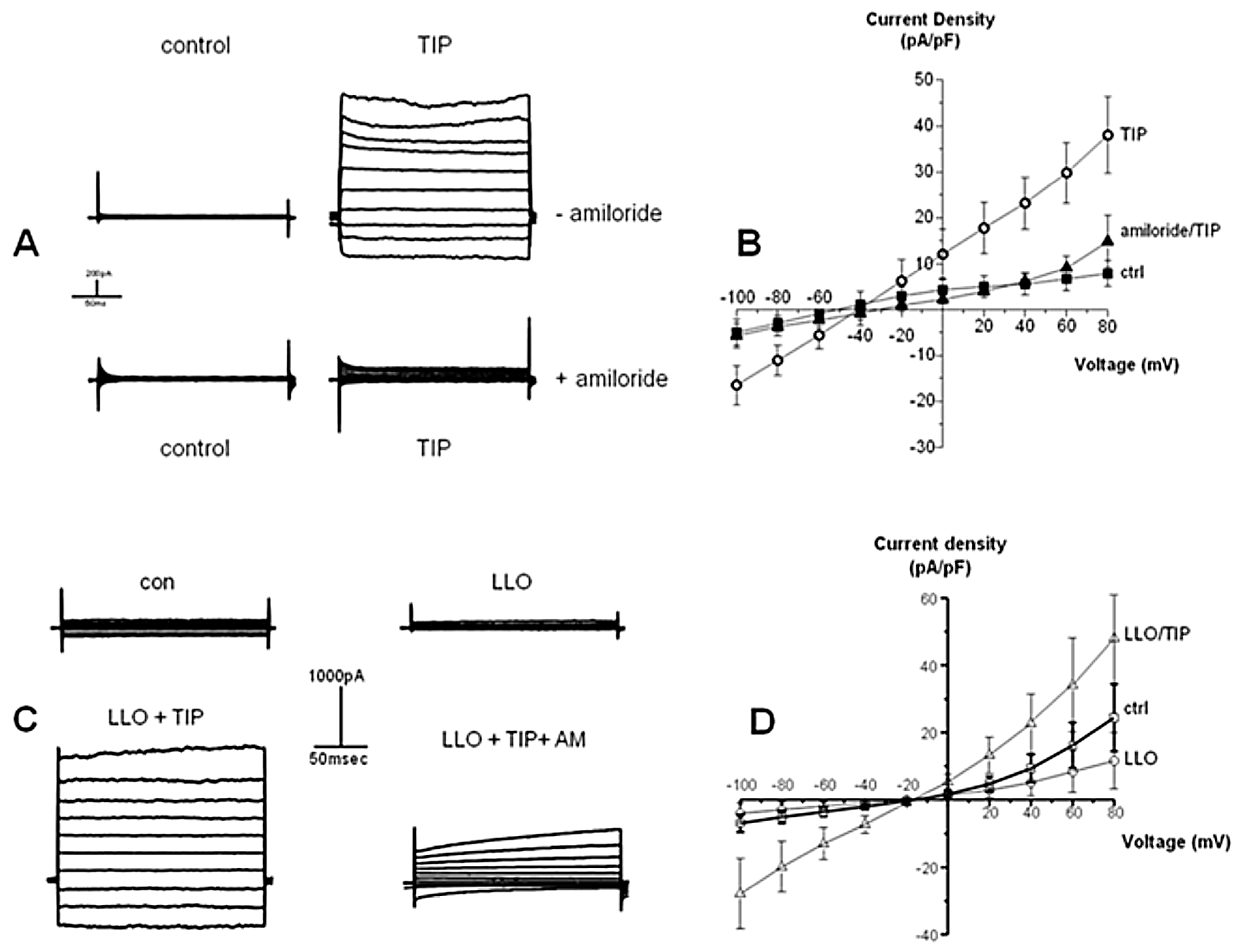
2.6. LLO Decreases Amiloride-Sensitive Sodium Current in H441 Cells
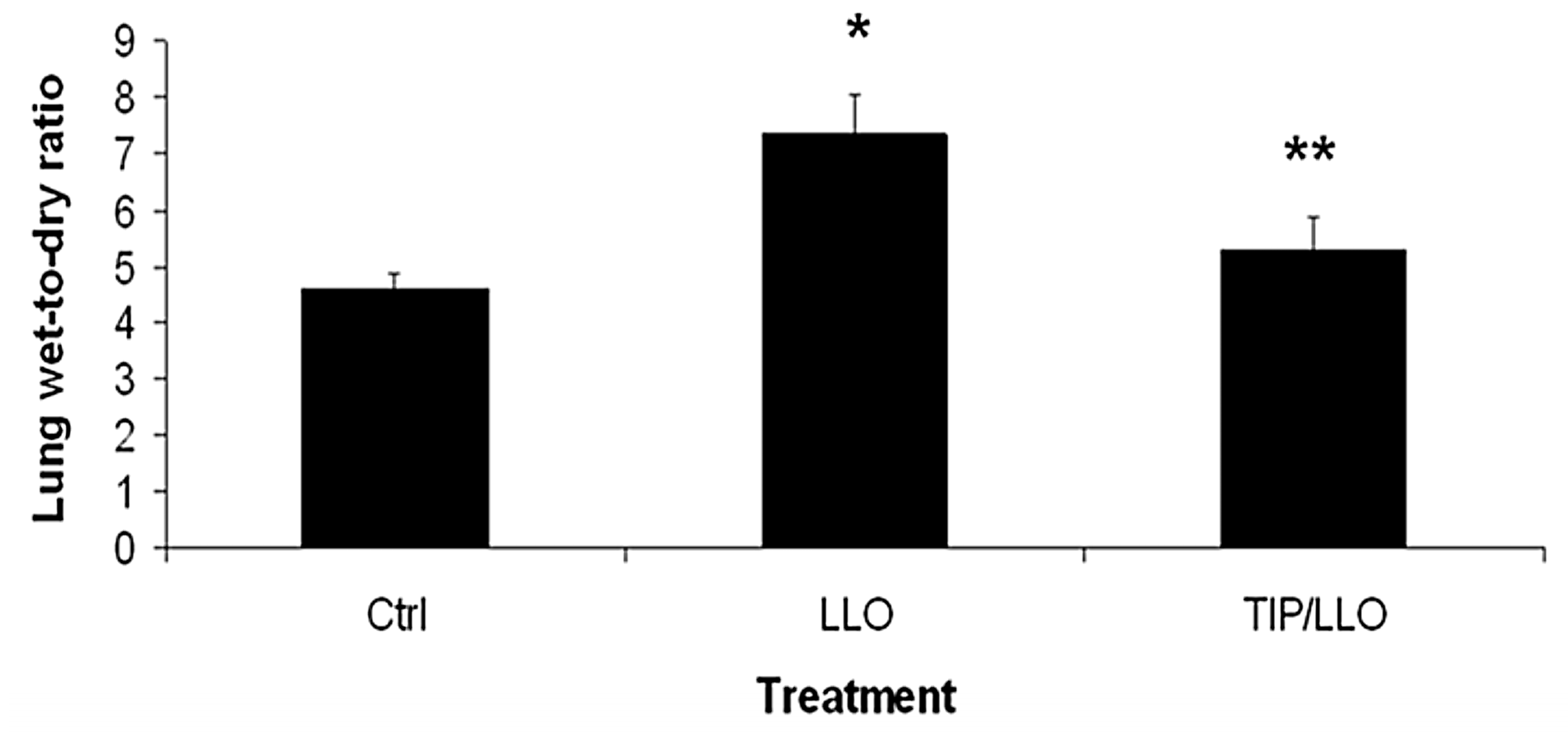
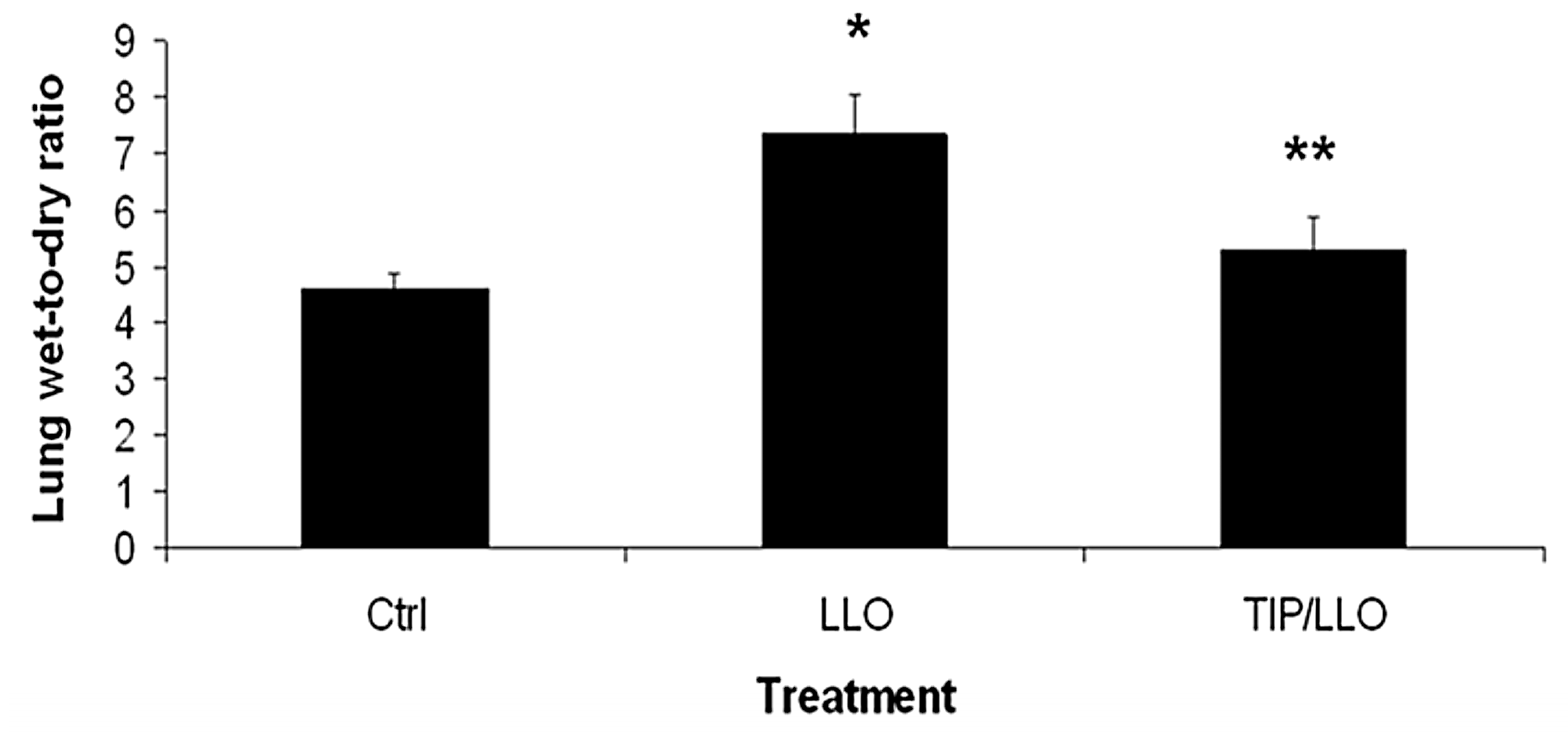
2.7. The TNF-Derived TIP Peptide Reduces Edema Formation in LLO-Treated C57BL/6 Mice
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cells
5.2. Animals
5.3. Peptides
- human TIP (hTIP) peptide: CGQRETPEGAEAKPWYC
- scrambled TIP peptide: CGTKPWEAGEQERPAYC
5.4. Chemicals and Antibodies
5.5. Purification of LLO
5.6. Immunoblotting
5.7. Surface Biotinylation
5.8. Measurement of PKC-α Activation in H441 Cells
5.9. Whole Cell Patch Clamp Recording
5.10. Determination of the Lung Wet-to-Dry Weight Ratio
5.11. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allerberger, F.; Wagner, M. Listeriosis: A resurgent foodborne infection. Clin. Microbiol. Infect. 2010, 16, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Ananthraman, A.; Israel, R.H.; Magnussen, C.R. Pleural-pulmonary aspects of Listeria monocytogenes infection. Respiration 1983, 44, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Lerolle, N.; Zahar, J.R.; Duboc, V.; Tissier, F.; Rabbat, A. Pneumonia involving Legionella pneumophila and Listeria monocytogenes in an immunocompromised patient: An unusual coinfection. Respiration 2002, 69, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Alouf, J.E. Pore-forming bacterial protein toxins: An overview. Curr. Top. Microbiol. Immunol. 2001, 257, 1–14. [Google Scholar] [PubMed]
- Repp, H.; Pamukçi, Z.; Koschinski, A.; Domann, E.; Darji, A.; Birringer, J.; Brockmeier, D.; Chakraborty, T.; Dreyer, F. Listeriolysin of Listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cell. Microbiol. 2002, 4, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.C.; Helms, M.N.; Koval, M.; Bao, H.F.; Jain, L. The Contribution of Epithelial Sodium Channels to Alveolar Function in Health and Disease. Annu. Rev. Physiol. 2009, 71, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Garty, H.; Palmer, L.G. Epithelial sodium channels: Function, structure, and regulation. Physiol. Rev. 1997, 77, 359–396. [Google Scholar] [CrossRef] [PubMed]
- Hummler, E.; Barker, P.; Gatzy, J.; Beermann, F.; Verdumo, C.; Schmidt, A.; Boucher, R.; Rossier, B.C. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat. Genet. 1996, 12, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.L.; Zhao, R.Z.; Chen, Z.X.; Shetty, S.; Idell, S.; Matalon, S. δ ENaC: A novel divergent amiloride-inhibitable sodium channel. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L1013–L1026. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Raviv, I.; Sznajder, J.I. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensiv. Care Med. 2007, 33, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Debonneville, C.; Flores, S.Y.; Kamynina, E.; Plant, P.J.; Tauxe, C.; Thomas, M.A.; Munster, C.; Chraibi, A.; Pratt, J.H.; Horisberger, J.D.; et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Bhargava, A.; Mastroberardino, L.; Meijer, O.C.; Wang, J.; Buse, P.; Firestone, G.L.; Verrey, F.; Pearce, D. Epithelial sodium channel regulated by aldosterone-induced protein SGK. Proc. Natl. Acad. Sci. USA 1999, 96, 2514–2519. [Google Scholar] [CrossRef] [PubMed]
- Naray-Fejes-Toth, A.; Canessa, C.; Cleaveland, E.S.; Aldrich, G.; Fejes-Toth, G. SGK is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial Na+ channels. J. Biol. Chem. 1999, 274, 16973–16978. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Dinudom, A.; Sanchez-Perez, A.; Kumar, S.; Cook, D.I. AKT mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J. Biol. Chem. 2007, 282, 29866–29873. [Google Scholar] [CrossRef] [PubMed]
- Diakov, A.; Korbmacher, C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s alpha-subunit. J. Biol. Chem. 2004, 279, 38134–38142. [Google Scholar] [CrossRef] [PubMed]
- Helms, M.N.; Yu, L.; Malik, B.; Kleinhenz, D.J.; Hart, C.M.; Eaton, D.C. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am. J. Physiol. Cell Physiol. 2005, 289, C717–C726. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, T.; Yamagata, Y.; Masse, C.; Tessier, M.C.; Brochiero, E.; Dagenais, A.; Berthiaume, Y. Modulation of Na+ transport and epithelial sodium channel expression by protein kinase C in rat alveolar epithelial cells. Can. Physiol. Pharmacol. 2005, 83, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.F.; Zhang, Z.R.; Liang, Y.Y.; Ma, J.J.; Eaton, D.C.; Ma, H.P. Ceramide mediates inhibition of the renal epithelial sodium channel by tumor necrosis factor-alpha through protein kinase C. Am. J. Physiol. 2007, 293, F1178–F1186. [Google Scholar]
- Kunzelmann, K.; Beesley, A.H.; King, N.J.; Karupiah, G.; Young, J.A.; Cook, D.I. Influenza virus inhibits amiloride-sensitive Na+ channels in respiratory epithelia. Proc. Natl. Acad. Sci. USA 2000, 97, 10282–10287. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Seth, S.; Yue, G.; Kamat, P.; Compans, R.W.; Guidot, D.; Brown, L.A.; Eaton, D.C.; Jain, L. Influenza virus inhibits ENaC and lung fluid clearance. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L366–L373. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.E.; Lazarowski, E.R.; Traylor, Z.P.; Yu, E.N.; Jewell, N.A.; Durbin, R.K.; Durbin, J.E.; Davis, I.C. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am. J. Respir. Crit. Care Med. 2008, 178, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Boncoeur, E.; Tardif, V.; Tessier, M.C.; Morneau, F.; Lavoie, J.; Gendreau-Berthiaume, E.; Grygorczyk, R.; Dagenais, A.; Berthiaume, Y. Modulation of epithelial sodium channel activity by lipopolysaccharide in alveolar type II cells: Involvement of purinergic signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L417–L426. [Google Scholar] [CrossRef] [PubMed]
- Sherblom, A.P.; Decker, J.M.; Muchmore, A.V. The lectin-like interaction between recombinant tumor necrosis factor and uromodulin. J. Biol. Chem. 1988, 263, 5418–5424. [Google Scholar] [PubMed]
- Lucas, R.; Magez, S.; De Leys, R.; Fransen, L.; Scheerlinck, J.P.; Rampelberg, M.; Sablon, E.; De Baetselier, P. Mapping the lectin-like activity of tumor necrosis factor. Science 1994, 263, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Daulouède, S.; Bouteille, B.; Moynet, D.; De Baetselier, P.; Courtois, P.; Lemesre, J.L.; Buguet, A.; Cespuglio, R.; Vincendeau, P. Human macrophage tumor necrosis factor (TNF)-alpha production induced by Trypanosoma brucei gambiense and the role of TNF-alpha in parasite control. J. Infect. Dis. 2001, 183, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Magez, S.; Geuskens, M.; Beschin, A.; del Favero, H.; Verschueren, H.; Lucas, R.; Pays, E.; de Baetselier, P. Specific uptake of tumor necrosis factor-alpha is involved in growth control of Trypanosoma brucei. J. Cell Biol. 1997, 137, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.A.C.; Alves, A.M.C.V.; Rocha, M.F.G.; Cordeiro, R.A.; Brilhante, R.S.N.; Pinto, A.C.M.D.; Nunes, R.M.; Girão, V.C.C.; Sidrim, J.J.C. Tumor necrosis factor prevents Candida albicans biofilm formation. Sci. Rep. 2017, 7, 1206. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Jayr, C.; Lazrak, A.; Wang, Y.; Lucas, R.; Matalon, S.; Matthay, M.A. Mechanisms of TNF-alpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1258–L1265. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.; Hamacher, J.; Morel, D.R.; Wendel, A.; Lucas, R. Dichotomal role of TNF in experimental pulmonary edema reabsorption. J. Immunol. 2005, 175, 3402–3408. [Google Scholar] [CrossRef] [PubMed]
- Elia, N.; Tapponnier, M.; Matthay, M.A.; Hamacher, J.; Pache, J.C.; Brundler, M.A.; Totsch, M.; De Baetselier, P.; Fransen, L.; Fukuda, N.; et al. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am. J. Respir. Crit. Care Med. 2003, 168, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Hamacher, J.; Stammberger, U.; Roux, J.; Kumar, S.; Yang, G.; Xiong, C.; Schmid, R.A.; Fakin, R.M.; Chakraborty, T.; Hossain, H.M.; et al. The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation—Potential role for a reduction in reactive oxygen species generation. Crit. Care Med. 2010, 38, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, I.; Schermuly, R.T.; Ghofrani, H.A.; Rummel, S.; Wehner, S.; Muhldorfer, I.; Schafer, K.P.; Seeger, W.; Morty, R.E.; Grimminger, F.; et al. The lectin-like domain of tumor necrosis factor-alpha improves alveolar fluid balance in injured isolated rabbit lungs. Crit. Care Med. 2008, 36, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Czikora, I.; Alli, A.; Bao, H.F.; Kaftan, D.; Sridhar, S.; Apell, H.J.; Gorshkov, B.; White, R.; Zimmermann, A.; Wendel, A.; et al. A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am. J. Respir. Crit. Care Med. 2014, 190, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Yue, Q.; Alli, A.; Duke, B.J.; Al-Khalili, O.; Thai, T.L.; Hamacher, J.; Sridhar, S.; Lebedyeva, I.; Su, H.; et al. The Lectin-like Domain of TNF Increases ENaC Open Probability through a Novel Site at the Interface between the Second Transmembrane and C-terminal Domains of the α-Subunit. J. Biol. Chem. 2016, 291, 23440–23451. [Google Scholar] [CrossRef] [PubMed]
- Alli, A.A.; Bao, H.F.; Aldrugh, Y.; Song, J.Z.; Ma, H.P.; Yu, L.; Al-Khalili, O.; Eaton, D.C. Phosphatidylinositol phosphate-dependent regulation of Xenopus ENaC by MARCKS protein. Am. J. Physiol. Ren. Physiol. 2012, 303, F800–F811. [Google Scholar] [CrossRef] [PubMed]
- Planès, C.; Randrianarison, N.H.; Charles, R.P.; Frateschi, S.; Cluzeaud, F.; Vuagniaux, G.; Soler, P.; Clerici, C.; Rossier, B.C.; Hummler, E. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol. Med. 2010, 2, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. 2007, 7, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.N.; Eaton, D.C. Effects of luminal Na+ on single Na+ channels in A6 cells, a regulatory role for protein kinase C. Am. J. Physiol. 1989, 256, F1094–F1103. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.M.; Kieloch, A.; Currie, R.A.; Deak, M.; Alessi, D.R. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001, 20, 4380–4390. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Zhou, Q.L.; Holik, J.; Patel, S.; Leszyk, J.; Coleman, K.; Chouinard, M.; Czech, M.P. Identification of WNK1 as a substrate of Akt/protein kinase B and a negative regulator of insulin-stimulated mitogenesis in 3T3-L1 cells. J. Biol. Chem. 2005, 280, 21622–21628. [Google Scholar] [CrossRef] [PubMed]
- Ramminger, S.J.; Richard, K.; Inglis, S.K.; Land, S.C.; Olver, R.E.; Wilson, S.M. A regulated apical Na(+) conductance in dexamethasone-treated H441 airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L411–L419. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.G.; Gallacher, M.; Olver, R.E.; Wilson, S.M. The regulation of selective and nonselective Na+ conductances in H441 human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L942–L954. [Google Scholar] [CrossRef] [PubMed]
- Taruno, A.; Niisato, N.; Marunaka, Y. Intracellular calcium plays a role as the second messenger of hypotonic stress in gene regulation of SGK1 and ENaC in renal epithelial A6 cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F177–F186. [Google Scholar] [CrossRef] [PubMed]
- Rezaiguia, S.; Garat, C.; Delclaux, C.; Meignan, M.; Fleury, J.; Legrand, P.; Matthay, M.A.; Jayr, C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J. Clin. Investig. 1997, 99, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, R.H.; Kapoor, N.; Qadri, Y.J.; Anderson, S.J.; Fuller, C.M.; Benos, D.J. Heteromeric assembly of acid-sensitive ion channel and epithelial sodium channel subunits. J. Biol. Chem. 2007, 282, 25548–25559. [Google Scholar] [CrossRef] [PubMed]
- Trac, P.T.; Thai, T.L.; Linck, V.; Zou, L.; Greenlee, M.M.; Yue, Q.; Al-Khalili, O.; Alli, A.A.; Eaton, A.F.; Eaton, D.C. Alveolar non-selective channels are ASIC1a/α-ENaC channels and contribute to AFC. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L797–L811. [Google Scholar] [CrossRef] [PubMed]
- Czikora, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front. Immunol. 2017, 8, 842. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Yang, G.; Kumar, S.; Aggarwal, S.; Leustik, M.; Snead, C.; Hamacher, J.; Fischer, B.; Umapathy, N.S.; Hossain, H.; et al. The lectin-like domain of TNF protects from listeriolysin-induced hyperpermeability in human pulmonary microvascular endothelial cells—A crucial role for protein kinase C-alpha inhibition. Vascul. Pharmacol. 2010, 52, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Krenn, K.; Lucas, R.; Croizé, A.; Boehme, S.; Klein, K.U.; Hermann, R.; Markstaller, K.; Ullrich, R. Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: A phase IIa randomized placebo-controlled trial. Crit. Care 2017, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Bartoszewski, R.; Collawn, J.F. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1229–L1238. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.C.; Malik, B.; Bao, H.F.; Yu, L.; Jain, L. Regulation of epithelial sodium channel trafficking by ubiquitination. Proc. Am. Thorac. Soc. 2010, 7, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, M.B.; Edinger, R.S.; Silvis, M.R.; Gallo, L.I.; Liang, X.; Apodaca, G.; Frizzell, R.A.; Johnson, J.P. Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC). Am. J. Physiol. Ren. Physiol. 2012, 302, F581–F590. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.A.; Batsche, E.; Regnault, B.; Tham, T.N.; Seveau, S.; Muchardt, C.; Cossart, P. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. USA 2007, 104, 13467–13472. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Hamon, M.; Gouin, E.; Nahori, M.A.; Impens, F.; Neyret-Kahn, H.; Gevaert, K.; Vandekerckhove, J.; Dejean, A.; Cossart, P. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 2010, 464, 1192–1195. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Pillich, H.; White, R.; Czikora, I.; Pochic, I.; Yue, Q.; Hudel, M.; Gorshkov, B.; Verin, A.; Sridhar, S.; et al. Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells. Toxins 2018, 10, 79. https://doi.org/10.3390/toxins10020079
Yang G, Pillich H, White R, Czikora I, Pochic I, Yue Q, Hudel M, Gorshkov B, Verin A, Sridhar S, et al. Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells. Toxins. 2018; 10(2):79. https://doi.org/10.3390/toxins10020079
Chicago/Turabian StyleYang, Guang, Helena Pillich, Richard White, Istvan Czikora, Isabelle Pochic, Qiang Yue, Martina Hudel, Boris Gorshkov, Alexander Verin, Supriya Sridhar, and et al. 2018. "Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells" Toxins 10, no. 2: 79. https://doi.org/10.3390/toxins10020079
APA StyleYang, G., Pillich, H., White, R., Czikora, I., Pochic, I., Yue, Q., Hudel, M., Gorshkov, B., Verin, A., Sridhar, S., Isales, C. M., Eaton, D. C., Hamacher, J., Chakraborty, T., & Lucas, R. (2018). Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells. Toxins, 10(2), 79. https://doi.org/10.3390/toxins10020079