A Balanced Risk–Benefit Analysis to Determine Human Risks Associated with Pyrrolizidine Alkaloids (PA)—The Case of Tea and Herbal Infusions

 ,
, 
Abstract
:1. Introduction
2. Health Effects and Safety of Consuming Tea and Herbal Infusions
Safety Assessment of Drinking Tea
3. PA Toxicity: Analytical Detection, Pathogenesis, and Etiology
3.1. Analytical Detection of PA
3.2. PA Induced Toxicity: Pathogenesis
3.3. PA Induced Toxicity: Etiology
4. PA Toxicity: How Do Experimental Models and Findings in Humans Fit Together?
4.1. Combination of Animal and Human Toxicity Data Demonstrate the Relevance of Analogous Manifestation of Toxicity
4.2. Epidemiological Evidence
5. PA Exposure in Humans: Main Sources, Ways to Avoid It, and Competitive Risks
5.1. Main Sources of Human PA Exposure
5.2. PA Contamination and Ways to Avoid and Control It
5.3. The Need to Consider Competitive Risks
6. Balanced Risk–Benefit and Risk Communication
6.1. Objective versus Subjective Risks
6.2. PA Fact Collection to Provide Reference Points for Personal Decision Making
7. Conclusions
Conflicts of Interest
References
- Wiedenfeld, H.; Roeder, E.; Bourauel, T.; Edgar, J. Pyrrolizidine Alkaloids. Structure and Toxicity; V&R Unipress: Göttingen, Germany, 2008. [Google Scholar]
- Bundesinstitut für Risikobewertung. Frequently Asked Questions on Pyrrolizidine Alkaloids in Foods. Available online: http://www.bfr.bund.de/cm/349/frequently-asked-questions-on-pyrrolizidine-alkaloids-in-foods.pdf (accessed on 11 May 2017).
- Merz, K.-H.; Schrenk, D. Interim relative potency factors for the toxicological risk assessment of pyrrolizidine alkaloids in food and herbal medicines. Toxicol. Lett. 2016, 263, 44–57. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Public statement on the Use of Herbal Medicinal Products Containing Toxic, Unsaturated Pyrrolizidine Alkaloids (PAs). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Public_statement/2014/12/WC500179559.pdf (accessed on 9 May 2017).
- Mulder, P.P.; Sánchez, P.L.; These, A.; Preiss-Weigert, A.; Castellari, M. Occurrence of Pyrrolizidine Alkaloids in food. EFS3 2015, 12. [Google Scholar] [CrossRef]
- Wiedenfeld, H. Plants containing pyrrolizidine alkaloids: Toxicity and problems. Food Addit. Contam. Part A 2011, 28, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Habs, M.; Habs, H.; Forth, W. Kanzerogene Naturprodukte: Risikobewertung pyrrolizidinhaltiger Arzneistoffe. Dtsch. Ärzteblatt 1991, 88, 3425–3432. [Google Scholar]
- Wiedenfeld, H.; Edgar, J. Toxicity of pyrrolizidine alkaloids to humans and ruminants. Phytochem. Rev. 2011, 10, 137–151. [Google Scholar] [CrossRef]
- Bundesinstitut für Risikobewertung. Vorläufige Empfehlungen des BfR zur Analytik von Pyrrolizidinalkaloiden (PA) in Kräutertee und Tee Analytspektrum und Probenahmeverfahren). Available online: http://www.bfr.bund.de/cm/343/vorlaeufige-empfehlungen-des-bfr-zur-analytik-von-pyrrolizidinalkaloiden-pa-in-kraeutertee-und-tee.pdf (accessed on 11 May 2017).
- European Medicines Agency. Committee on Herbal Medicinal Products (HMPC). Available online: www.ema.europa.eu>home>committees>HMPC>Overview (accessed on 1 September 2016).
- Edwards, S.E.; Costa Rocha, I.D.; Williamson, E.M.; Heinrich, M. Phytopharmacy. An Evidence-Based Guide to Herbal Medicinal Products; Wiley Blackwell: Chichester, UK, 2015. [Google Scholar]
- Wiedenfeld, H. Aufnahmewege von PA Durch Direkte und Indirekte Intoxikation. Available online: https://www.ak-kreuzkraut.de/toxizität-mensch-tier/humangefährdung/aufnahmewege/ (accessed on 11 May 2017).
- Pawar, R.S. Pyrrolizidine alkaloids. In Bad Bug Book: Foodborne Pathogenic Microorganisms and Natural Toxins; Food and Drug Administration: Silver Spring, MD, USA, 2012; pp. 242–244. [Google Scholar]
- Rasenack, R.; Muller, C.; Kleinschmidt, M.; Rasenack, J.; Wiedenfeld, H. Veno-occlusive disease in a fetus caused by pyrrolizidine alkaloids of food origin. Fetal Diagn. Ther. 2003, 18, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Statistical Portal. Annual Per Capita Tea Consumption Worldwide as of 2016, by Leading Countries (In Pounds). Available online: https://www.statista.com/statistics/507950/global-per-capita-tea-consumption-by-country/ (accessed on 6 October 2016).
- Khan, N.; Mukhtar, H. Tea and health: Studies in humans. Curr. Pharm. Des. 2013, 19, 6141–6147. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Rio, D.; N’Dri, Y.; Bettuzzi, S.; Peluso, I. Health Benefits of Tea. In Herbal Medicine; Wachtel-Galor, S., Ed.; Oxidative Stress and Disease; CRC Press: Boca Raton, FL, USA, 2011; pp. 239–261. [Google Scholar]
- Farzaneh, V.; Carvalho, I.S. A review of the health benefit potentials of herbal plant infusions and their mechanism of actions. Ind. Crops Prod. 2015, 65, 247–258. [Google Scholar] [CrossRef]
- Hartley, L.; Flowers, N.; Holmes, J.; Clarke, A.; Stranges, S.; Hooper, L.; Rees, K. Green and black tea for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Boehm, K.; Borrelli, F.; Ernst, E.; Habacher, G.; Hung, S.K.; Milazzo, S.; Horneber, M. Green tea (Camellia sinensis) for the prevention of cancer. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Chen, Z.-M.; Lin, Z. Tea and human health: Biomedical functions of tea active components and current issues. J. Zhejiang Univ. Sci. B 2015, 16, 87–102. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Tea and Cancer Prevention. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/diet/tea-fact-sheet (accessed on 11 May 2017).
- Jin, X.; Zheng, R.-H.; Li, Y.-M. Green tea consumption and liver disease: A systematic review. Liver Int. 2008, 28, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Yang, J.; Li, T.; Song, L.; Han, T.; Yang, M.; Liao, H.; He, J.; Zhong, X. The effect of green tea intake on risk of liver disease: A meta analysis. Int. J. Clin. Exp. Med. 2015, 8, 8339–8346. [Google Scholar] [PubMed]
- Dong, X.; Yang, C.; Cao, S.; Gan, Y.; Sun, H.; Gong, Y.; Yang, H.; Yin, X.; Lu, Z. Tea consumption and the risk of depression: A meta-analysis of observational studies. Aust. N. Z. J. Psychiatr. 2015, 49, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-S.; Wang, W.-Y.; Fan, W.-Y.; Deng, Q.; Wang, X. Tea consumption and risk of type 2 diabetes: A dose–response meta-analysis of cohort studies. Br. J. Nutr. 2014, 111, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Qin, Y.-Y.; Wei, X.; Yu, F.-F.; Zhou, Y.-H.; He, J. Tea consumption and risk of cardiovascular outcomes and total mortality: A systematic review and meta-analysis of prospective observational studies. Eur. J. Epidemiol. 2015, 30, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Sakuae, M.; Reid, D. Making Tea in Place: Experiences of Women Engaged in a Japanese Tea Ceremony. J. Occup. Sci. 2012, 19, 283–291. [Google Scholar] [CrossRef]
- Steptoe, A.; Gibson, E.L.; Vuononvirta, R.; Williams, E.D.; Hamer, M.; Rycroft, J.A.; Erusalimsky, J.D.; Wardle, J. The effects of tea on psychophysiological stress responsivity and post-stress recovery: A randomised double-blind trial. Psychopharmacology 2007, 190, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tea & Herbal Infusions Europe. THIE Inventory List of Herbals Considered as Food. Available online: http://www.thie-online.eu/fileadmin/inhalte/Publications/HFI/2016/2016-06-24_PU_THIE_Inventory_List_of_Herbals_Considered_as_Food_final.pdf (accessed on 11 May 2017).
- Deutscher Teeverband e.V. Inlandskonsum auf Allzeithoch: Tee in Deutschland Beliebter denn je. Available online: http://www.teeverband.de/wirtschaft/pdf/2016-05-26_PM_WFT.pdf (accessed on 18 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. European Union Herbal Monograph on Matricaria Recutita L., Flos. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Herbal_monograph/2016/04/WC500204299.pdf (accessed on 15 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. Community Herbal Monograph on Cinnamomum Verum J.S. Presl, Cortex. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Community_herbal_monograph/2011/08/WC500110095.pdf (accessed on 15 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. Community Herbal Monograph on Foeniculum Vulgare Miller Subsp. Vulgare Var. Vulgare, Fructus. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Community_herbal_monograph/2009/12/WC500018464.pdf (accessed on 15 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. Community Herbal Monograph on Zingiber Officinale Roscoe, Rhizoma. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Community_herbal_monograph/2012/06/WC500128142.pdf (accessed on 15 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. Community Herbal Monograph on Melissa Officinalis L., Folium. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Community_herbal_monograph/2013/08/WC500147189.pdf (accessed on 15 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. Community Herbal Monograph on Rosmarinus Officinalis L., Aetheroleum. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Community_herbal_monograph/2011/01/WC500101493.pdf (accessed on 15 May 2017).
- European Medicines Agency; Committee on Herbal Medicinal Product. European Union Herbal Monograph on Valeriana Officinalis L., Radix. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Herbal_-_Herbal_monograph/2016/04/WC500205376.pdf (accessed on 12 December 2016).
- Smith, A. Effects of caffeine on human behavior. Food Chem. Toxicol. 2002, 40, 1243–1255. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA) Panel on Dietetic Products, Nutrition and Allergies (EFSA NDA Panel). Scientific Opinion on the safety of caffeine. EFSA J. 2015, 13, 4102. [Google Scholar] [CrossRef]
- Jurgens, T.M.; Whelan, A.M.; Killian, L.; Doucette, S.; Kirk, S.; Foy, E. Green tea for weight loss and weight maintenance in overweight or obese adults. Cochrane Database Syst. Rev. 2012, 12, CD008650. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on Pyrrolizidine alkaloids in food and feed. EFSA J. 2011, 9, 2406. [Google Scholar] [CrossRef]
- Crews, C.; Berthiller, F.; Krska, R. Update on analytical methods for toxic pyrrolizidine alkaloids. Anal. Bioanal. Chem. 2010, 396, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Nijs, M.D.; Elbers, I.J.W.; Mulder, P.P.J. Inter-laboratory comparison study for pyrrolizidine alkaloids in animal feed using spiked and incurred material. Food Addit. Contam. Part A 2014, 31, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Bodi, D.; Pydde, Y.; Preiß-Weigert, A. Internationale Laborvergleichsuntersuchung zur Bestimmung von Pyrrolizidinalkaloiden in Kräutertee und Rooibostee; BfR-Wissenschaft 2016, 3; BfR: Berlin, Germany, 2017. [Google Scholar]
- Committee on Herbal Medicinal Product. Guideline on Good Agricultural and Collecting Practice (GACP) for Starting Material of Herbal Origin. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003362.pdf (accessed on 11 May 2017).
- The Commission of the European Communities. COMMISSION REGULATION (EC) No. 401/2006: Of 23 February 2006 Laying Down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:070:0012:0034:EN:PDF (accessed on 11 May 2017).
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Ito, Y.; Bethea, N.W.; McCuskey, M.K.; Wang, X.; McCuskey, R.S. Embolization by sinusoidal lining cells obstructs the microcirculation in rat sinusoidal obstruction syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G1045–G1052. [Google Scholar] [CrossRef] [PubMed]
- Helmy, A. Review article: Updates in the pathogenesis and therapy of hepatic sinusoidal obstruction syndrome. Aliment. Pharmacol. Ther. 2006, 23, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Yang, M.; Fu, P.; Ye, Y.; Lin, G. Metabolic activation of pyrrolizidine alkaloids: Insights into the structural and enzymatic basis. Chem. Res. Toxicol. 2014, 27, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Storer, R.D.; Criswell, K.A.; Doerrer, N.G.; Dellarco, V.L.; Pegg, D.G.; Wojcinski, Z.W.; Malarkey, D.E.; Jacobs, A.C.; Klaunig, J.E.; et al. Hemangiosarcoma in rodents: Mode-of-action evaluation and human relevance. Toxicol. Sci. 2009, 111, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Mei, N.; Xia, Q.; Chen, T.; Chan, P.-C.; Fu, P.P. Gene expression profiling as an initial approach for mechanistic studies of toxicity and tumorigenicity of herbal plants and herbal dietary supplements. J. Environ. Sci. Health Part C 2010, 28, 60–87. [Google Scholar] [CrossRef] [PubMed]
- Luckert, C.; Hessel, S.; Lenze, D.; Lampen, A. Disturbance of gene expression in primary human hepatocytes by hepatotoxic pyrrolizidine alkaloids: A whole genome transcriptome analysis. Toxicology 2015, 29, 1669–1682. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Holsapple, M.P.; Pitot, H.C.; Cohen, S.M.; Boobis, A.R.; Klaunig, J.E.; Pastoor, T.; Dellarco, V.L.; Dragan, Y.P. Mode of action in relevance of rodent liver tumors to human cancer risk. Toxicol. Sci. 2006, 89, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, T.; Gaus, W. Cross matching observations on toxicological and clinical data for the assessment of tolerability and safety of Ginkgo biloba leaf extract. Toxicology 2015, 327, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Nnane, I.P.; Cheng, T.-Y. The effects of pretreatment with glycyrrhizin and glycyrrhetinic acid on the retrorsine-induced hepatotoxicity in rats. Toxicon 1999, 37, 1259–1270. [Google Scholar] [CrossRef]
- DeLeve, L.D.; McCuskey, R.S.; Wang, X.; Hu, L.; McCuskey, M.K.; Epstein, R.B.; Kanel, G.C. Characterization of a reproducible rat model of hepatic veno-occlusive disease. Hepatology 1999, 29, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Public Statement on Contamination of Herbal Medicinal Products/traditional Herbal Medicinal Products with Pyrrolizidine Alkaloids: Transitional Recommendations for Risk Managementand Quality Control. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Public_statement/2016/06/WC500208195.pdf (accessed on 9 May 2017).
- Kakar, F.; Akbarian, Z.; Leslie, T.; Mustafa, M.L.; Watson, J.; van Egmond, H.P.; Omar, M.F.; Mofleh, J. An outbreak of hepatic veno-occlusive disease in Western Afghanistan associated with exposure to wheat flour contaminated with pyrrolizidine alkaloids. J. Toxicol. 2010, 2010, 313280. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.P.; Yang, Y.-C.; Xia, Q.; Chou, M.W.; Cui, Y.Y.; Lin, G. Pyrrolizidine Alkaloids: Tumorigenic Components in Chinese Herbal Medicines and Dietary Supplements. J. Food Drug Anal. 2002, 10, 198–211. [Google Scholar]
- Habs, H.; Habs, M.; Marquardt, H.; Roder, E.; Schmahl, D.; Wiedenfeld, H. Carcinogenic and mutagenic activity of an alkaloidal extract of Senecio nemorensis ssp. fuchsii. Arzneim. Forsch. 1982, 32, 144–148. [Google Scholar]
- Heywwod, R. Clinical toxicity—Could it have been predicted? Post-marketing experience. In Proceedings of the Animal Toxicity Studies: Their Relevance for Man, London, UK, 26 September 1989; pp. 57–67. [Google Scholar]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Intentional Human Dosing Studies for EPA Regulatory Purposes. Scientific and Ethical Issues; National Academies Press: Washington, DC, USA, 2004. [Google Scholar]
- Ferdowsian, H.R.; Beck, N. Ethical and scientific considerations regarding animal testing and research. PLoS ONE 2011, 6, e24059. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A. The flaws and human harms of animal experimentation. Camb. Q. Healthc. Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Radiation; A Review of Human Carcinogens, 100D; IARC: Lyon, France, 2012. [Google Scholar]
- Schultheiss, P.C. A retrospective study of visceral and nonvisceral hemangiosarcoma and hemangiomas in domestic animals. J. Vet. Diagn. Investig. 2004, 16, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.A.; Trapp, S.; Phang, T.L.; Schappa, J.T.; Hunter, L.E.; Modiano, J.F. Gene expression profiles of sporadic canine hemangiosarcoma are uniquely associated with breed. PLoS ONE 2009, 4, e5549. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.B.; Winston, J.M.; Lee, C.C. Hepatic angiosarcoma: Animal model: Angiosarcoma of rats and mice induced by vinyl chloride. Am. J. Pathol. 1980, 101, 737–740. [Google Scholar] [PubMed]
- Adler, S.; Basketter, D.; Creton, S.; Pelkonen, O.; van Benthem, J.; Zuang, V.; Andersen, K.E.; Angers-Loustau, A.; Aptula, A.; Bal-Price, A.; et al. Alternative (non-animal) methods for cosmetics testing: Current status and future prospects-2010. Arch. Toxicol. 2011, 85, 367–485. [Google Scholar] [CrossRef] [PubMed]
- Basketter, D. A roadmap for the development of alternative (non-animal) methods for systemic toxicity testing. ALTEX 2012, 29, 3–91. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf (accessed on 18 May 2017).
- World Cancer Research Fund International. Cancer Facts & Figures: Worldwide Data. Available online: http://www.wcrf.org/int/cancer-facts-figures/worldwide-data (accessed on 15 May 2017).
- American Cancer Society. Cancer Facts & Figures 2016; American Cancer Society: Atlanta, GR, USA, 2016. [Google Scholar]
- Centers for Disease Control and Prevention. Liver Cancer. Available online: https://www.cdc.gov/cancer/liver/ (accessed on 15 May 2017).
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-P.; Robien, K.; Wang, R.; Govindarajan, S.; Yuan, J.-M.; Yu, M.C. Smoking as an independent risk factor for hepatocellular carcinoma: The Singapore Chinese Health Study. Br. J. Cancer 2011, 105, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Wambui, J.M.; Karuri, E.G.; Ojiambo, J.A.; Njage, P.M.K. Application of Probabilistic Modeling to Quantify the Reduction Levels of Hepatocellular Carcinoma Risk Attributable to Chronic Aflatoxins Exposure. Nutr. Cancer 2017, 69, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Ribes, J.; Diaz, M.; Cleries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127 (Suppl. 1), S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-Medicare database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.; Gogineni, V.; Saeian, K. Epidemiology of primary and secondary liver cancers. Semin. Interv. Radiol. 2006, 23, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.S.; Pereira, T.N.; Reilly, P.E.; Seawright, A.A. Pyrrolizidine alkaloids in human diet. Mutat. Res. 1999, 443, 53–67. [Google Scholar] [CrossRef]
- Falk, H.; Herbert, J.; Crowley, S.; Ishak, K.G.; Thomas, L.B.; Popper, H.; Caldwell, G.G. Epidemiology of hepatic angiosarcoma in the United States: 1964–1974. Environ. Health Perspect. 1981, 41, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.-C.; Wann, S.-R.; Chang, H.-T.; Lin, S.-L.; Wang, J.-S.; Guo, H.-R. Arsenic, vinyl chloride, viral hepatitis, and hepatic angiosarcoma: A hospital-based study and review of literature in Taiwan. BMC Gastroenterol. 2011, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Epidemiologic notes and reports: Angiosarcoma of the liver among polyvinyl chloride workers—Kentucky. Morb. Mortal. Wkly. Rep. 1997, 46, 97–101. [Google Scholar]
- Mani, H.; van Thiel, D.H. Mesenchymal tumors of the liver. Clin. Liver Dis. 2001, 5, 219–257. [Google Scholar] [CrossRef]
- Zocchetti, C. Angiosarcoma del fegato nell'uomo: Considerazioni epidemiologiche. La Medicina del lavoro 2001, 92, 39–53. [Google Scholar] [PubMed]
- Van Kampen, R.J.W.; Erdkamp, F.L.G.; Peters, F.P.J. Thorium dioxide-related haemangiosarcoma of the liver. Neth. J. Med. 2007, 65, 279–282. [Google Scholar] [PubMed]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Chemical Agents and Related Occupations; A Review of Human Carcinogens, 100F; IARC: Lyon, France, 2012. [Google Scholar]
- Chiou, H.Y.; Hsueh, Y.M.; Liaw, K.F.; Horng, S.F.; Chiang, M.H.; Pu, Y.S.; Lin, J.S.; Huang, C.H.; Chen, C.J. Incidence of internal cancers and ingested inorganic arsenic: A seven-year follow-up study in Taiwan. Cancer Res. 1995, 55, 1296–1300. [Google Scholar] [PubMed]
- Falk, H.; Thomas, L.B.; Popper, H.; Ishak, K.G. Hepatic angiosarcoma associated with androgenic-anabolic steroids. Lancet 1979, 2, 1120–1123. [Google Scholar] [CrossRef]
- Bodi, D.; Ronczka, S.; Gottschalk, C.; Behr, N.; Skibba, A.; Wagner, M.; Lahrssen-Wiederholt, M.; Preiss-Weigert, A.; These, A. Determination of pyrrolizidine alkaloids in tea, herbal drugs and honey. Food Addit. Contam. Part A 2014, 31, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Isomura, T.; Suzuki, S.; Origasa, H.; Hosono, A.; Suzuki, M.; Sawada, T.; Terao, S.; Muto, Y.; Koga, T. Liver-related safety assessment of green tea extracts in humans: A systematic review of randomized controlled trials. Eur. J. Clin. Nutr. 2016, 70, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). Dietary exposure assessment to pyrrolizidine alkaloids in the European population. EFSA J. 2016, 14, 1–50. [Google Scholar] [CrossRef]
- World Health Organization. WHO Guidelines on Good Agricultural and Collection Practices (GACP) for Medicinal Plants. Available online: http://apps.who.int/iris/bitstream/10665/42783/1/9241546271.pdf (accessed on 1 April 2017).
- Green, J.M. The benefits of herbicide-resistant crops. Pest Manag. Sci. 2012, 68, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Biber, P.; Weiss, U.; Dorna, M.; Albert, A. Navigation System of the Autonomous Agricultural Robot “BoniRob”. In Proceedings of the Workshop on Agricultural Robotics: Enabling Safe, Efficient, and Affordable Robots for Food Production, Algarve, Portugal, 11 October 2012. [Google Scholar]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.F.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Viral Hepatitis. Available online: http://www.cdc.gov/hepatitis (accessed on 18 May 2017).
- O’Connor, L.; Imamura, F.; Lentjes, M.A.H.; Khaw, K.-T.; Wareham, N.J.; Forouhi, N.G. Prospective associations and population impact of sweet beverage intake and type 2 diabetes, and effects of substitutions with alternative beverages. Diabetologia 2015, 58, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2014: Estimates of Diabetes and Its Burden in the United States; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014. [Google Scholar]
- Myers, J.P.; Antoniou, M.N.; Blumberg, B.; Carroll, L.; Colborn, T.; Everett, L.G.; Hansen, M.; Landrigan, P.J.; Lanphear, B.P.; Mesnage, R.; et al. Concerns over use of glyphosate-based herbicides and risks associated with exposures: A consensus statement. Environ. Health 2016, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Gigerenzer, G. Why does framing influence judgment? J. Gen. Intern. Med. 2003, 18, 960–961. [Google Scholar] [CrossRef] [PubMed]
- Spiegelhalter, D.J. Understanding uncertainty. Ann. Fam. Med. 2008, 6, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Spiegelhalter, D.; Pearson, M.; Short, I. Visualizing uncertainty about the future. Science 2011, 333, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Statistisches Bundesamt (Destatis). Gesundheit: Todesursachen in Deutschland 2015. Available online: https://www.destatis.de/DE/Publikationen/Thematisch/Gesundheit/Todesursachen/Todesursachen.html (accessed on 1 April 2017).
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems, 10th Revision, 2010th ed.; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Altman, D.G.; Bland, J.M. Statistics notes: Absence of evidence is not evidence of absence. BMJ 1995, 311, 485. [Google Scholar] [CrossRef] [PubMed]
- McDowell, M.; Rebitschek, F.G.; Gigerenzer, G.; Wegwarth, O. A Simple Tool for Communicating the Benefits and Harms of Health Interventions. MDM Policy Pract. 2016, 1. [Google Scholar] [CrossRef]
- Mukamal, K.J.; Maclure, M.; Muller, J.E.; Sherwood, J.B.; Mittleman, M.A. Tea consumption and mortality after acute myocardial infarction. Circulation 2002, 105, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Tea consumption and ovarian cancer risk in a population-based cohort. Arch. Intern. Med. 2005, 165, 2683–2686. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Titus-Ernstoff, L.; Newcomb, P.A.; Trentham-Dietz, A.; Anic, G.; Egan, K.M. Tea consumption and risk of breast cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Le, J.; Xie, L.P.; Lee, A.H.; Binns, C.W. Protective effect of green tea against prostate cancer: A case-control study in southeast China. Int. J. Cancer 2004, 108, 130–135. [Google Scholar] [CrossRef] [PubMed]
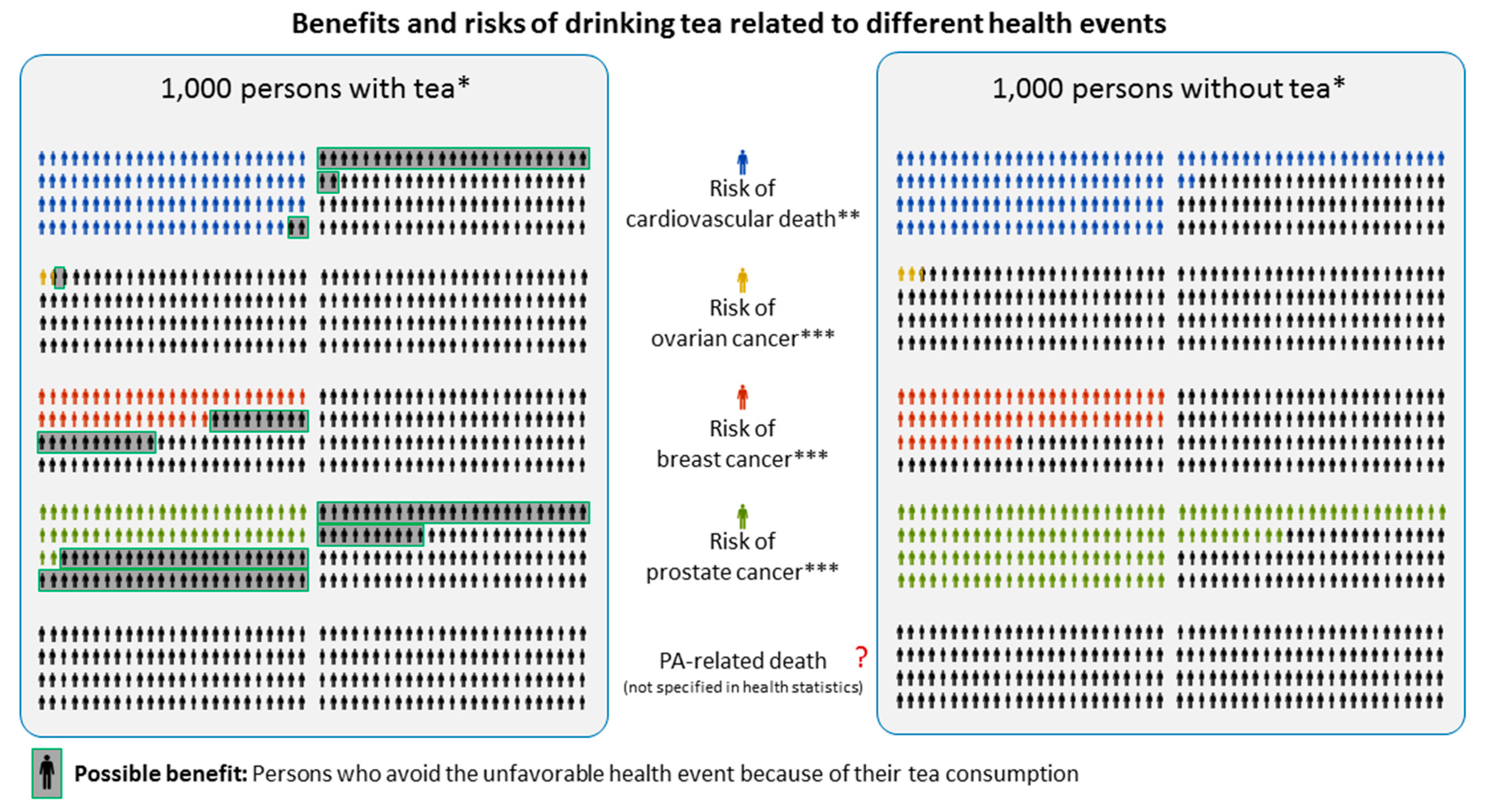

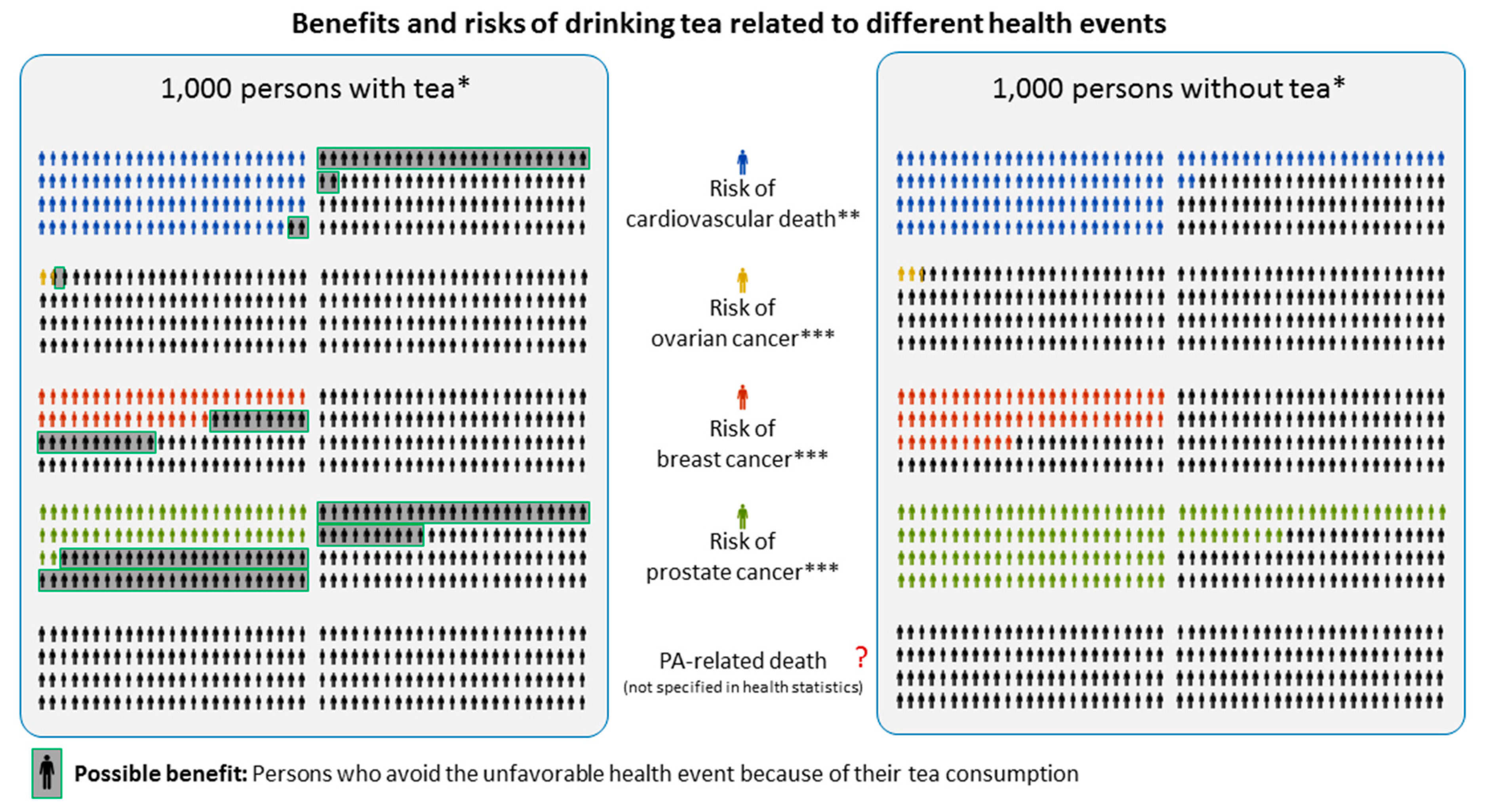{kind=link}
| Herb | Indications | Evidence | Monograph |
|---|---|---|---|
| Chamomilla (Matricaria chamomilla) | digestive ailment, bloating, flatulence, restlessness, mild insomnia | anecdotal, traditional use | [33] |
| Cinnamon (Cinnamomum verum) | digestive ailment, mild diarrhea | anecdotal, traditional use | [34] |
| Fennel (Foeniculum vulgare) | dyspepsia, spasmodic ailments | traditional use | [11,35] |
| Ginger (Zingiber officinale) | dyspepsia, flatulence, prevention of nausea, vomiting, motion sickness | well established and traditional use | [36] |
| Lemon balm (Melissa officinalis) | mild symptoms of stress, anxiety and insomnia | traditional use | [37] |
| Nettle (Urtica dioica) | lower urinary tract symptoms related to benign prostatic hyperplasia | traditional use | [36] |
| Peppermint (Mentha piperita) | digestive disorders, nausea, abdominal pain | traditional use | [11] |
| Rosemary (Rosmarinus officinalis) | dyspepsia, mild gastrointestinal spasmodic disorders | traditional use | [38] |
| Valerian (Valeriana officinalis) | mild nervous tension and sleep disorders | traditional use | [39] |
| Causes of Death Related to 1,000,000 Fatalities in Germany [108] | |
|---|---|
| Cardiovascular disease | 385,000 |
| Cancer | 250,000 |
| Diabetes mellitus | 26,000 |
| Liver diseases | 16,000 |
| Age between 60 and 65 | 11,000 |
| Household accidents | 10,000 |
| Liver cancer | 8500 |
| Car accidents | 3800 |
| Obesity | 2200 |
| Poisoning through pharmaceutical drugs | 2000 |
| Liver cancer (unexplained cause) | 1700 |
| Viral hepatitis | 1000 |
| Accidental poisoning | 750 |
| Unknown cause of death | 270 |
| Chronic hepatitis | 20 |
| PA-related death * | ? |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habs, M.; Binder, K.; Krauss, S.; Müller, K.; Ernst, B.; Valentini, L.; Koller, M. A Balanced Risk–Benefit Analysis to Determine Human Risks Associated with Pyrrolizidine Alkaloids (PA)—The Case of Tea and Herbal Infusions. Nutrients 2017, 9, 717. https://doi.org/10.3390/nu9070717
Habs M, Binder K, Krauss S, Müller K, Ernst B, Valentini L, Koller M. A Balanced Risk–Benefit Analysis to Determine Human Risks Associated with Pyrrolizidine Alkaloids (PA)—The Case of Tea and Herbal Infusions. Nutrients. 2017; 9(7):717. https://doi.org/10.3390/nu9070717
Chicago/Turabian StyleHabs, Michael, Karin Binder, Stefan Krauss, Karolina Müller, Brigitte Ernst, Luzia Valentini, and Michael Koller. 2017. "A Balanced Risk–Benefit Analysis to Determine Human Risks Associated with Pyrrolizidine Alkaloids (PA)—The Case of Tea and Herbal Infusions" Nutrients 9, no. 7: 717. https://doi.org/10.3390/nu9070717
APA StyleHabs, M., Binder, K., Krauss, S., Müller, K., Ernst, B., Valentini, L., & Koller, M. (2017). A Balanced Risk–Benefit Analysis to Determine Human Risks Associated with Pyrrolizidine Alkaloids (PA)—The Case of Tea and Herbal Infusions. Nutrients, 9(7), 717. https://doi.org/10.3390/nu9070717