Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism

{kind=link}
Abstract
:1. Introduction
2. Fructose and Hepatic Steatosis
2.1. Fructose as a Substrate of Hepatic de novo Lipogenesis
2.2. Fructose as an Inducer of De Novo Lipogenesis
3. Fructose and Disease Progression
3.1. Fructose and Oxidative Stress
3.2. Fructose and Endoplasmic Reticulum Stress
3.3. Fructose and Inflammation
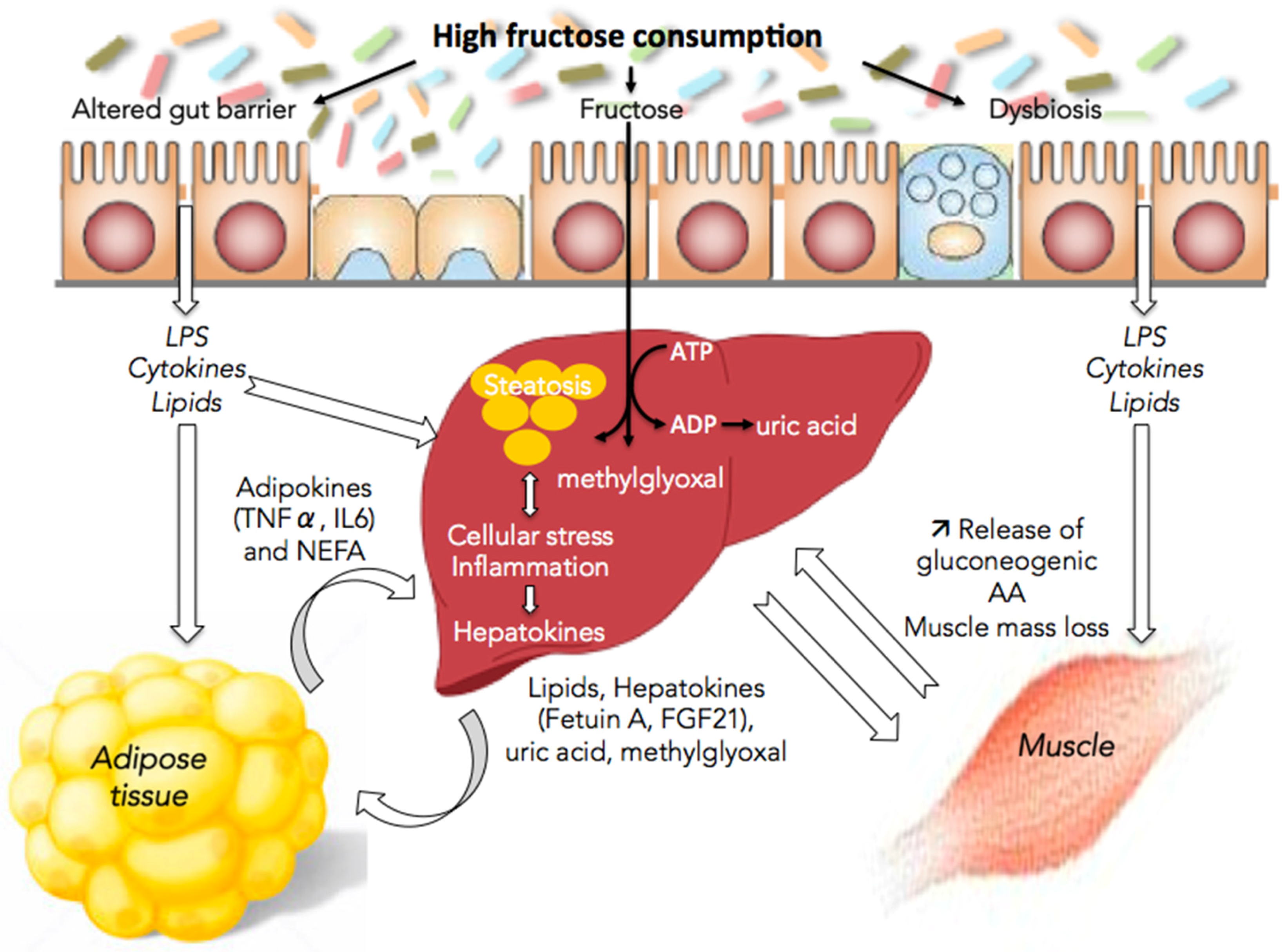
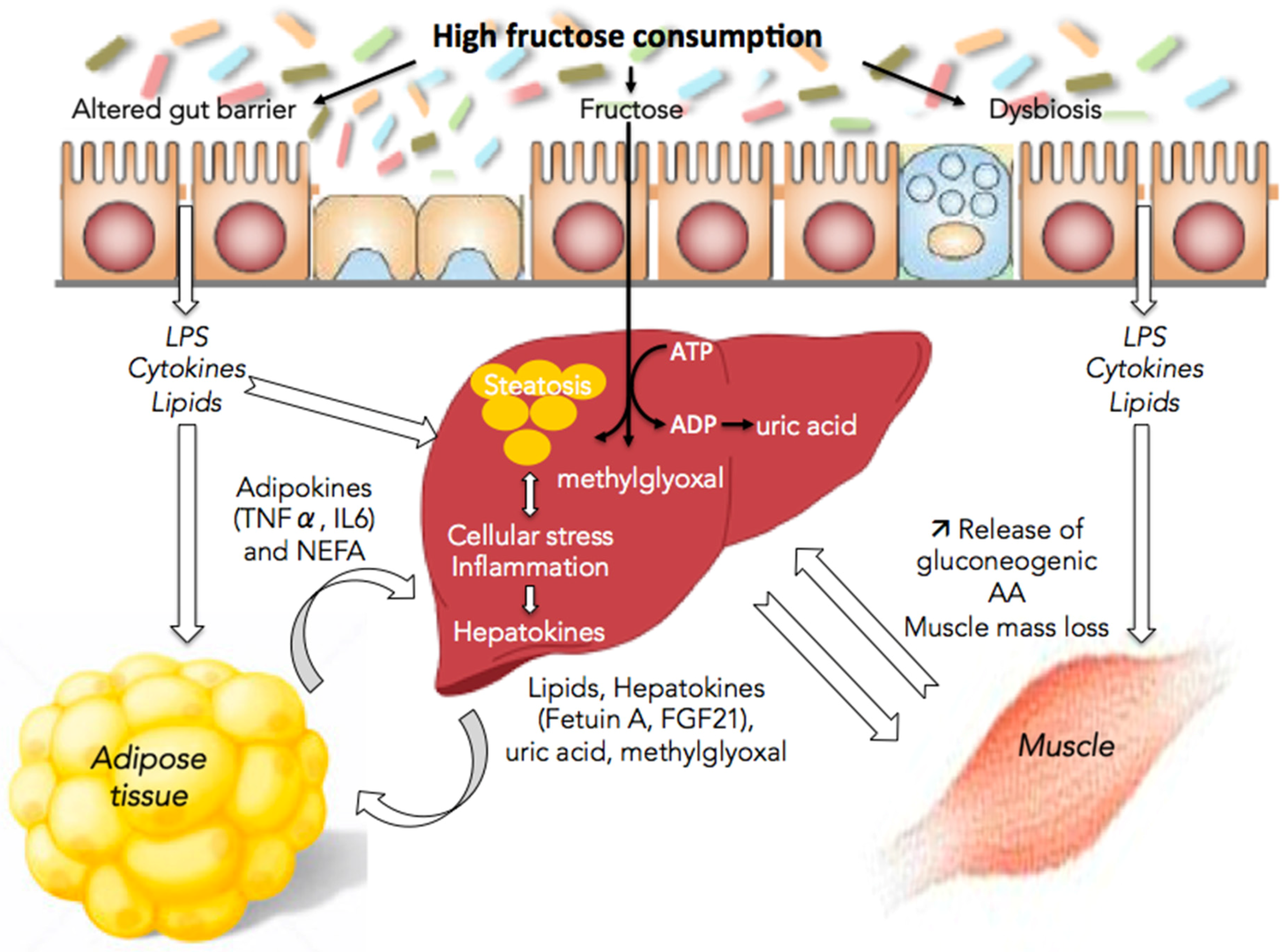
4. Fructose and Interorgan Cross-Talks
4.1. Fructose and the Gut/Liver Axis
4.2. Fructose and Adipose Tissue/Liver Axis
4.3. Fructose and Muscle/Liver Axis
5. Specific or Indirect Effect of Fructose
6. Fructose and NAFLD Management
7. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
References
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.L.; Leung, J.C.-F.; Loong, T.C.-W.; Wong, G.L.-H.; Yeung, D.K.-W.; Chan, R.S.-M.; Chan, H.L.; Chim, A.M.; Woo, J.; Chu, W.C.; et al. Prevalence and severity of nonalcoholic fatty liver disease in non-obese patients: A population study using proton-magnetic resonance spectroscopy. Am. J. Gastroenterol. 2015, 110, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [PubMed]
- Bray, G.A.; Popkin, B.M. Dietary sugar and body weight: Have we reached a crisis in the epidemic of obesity and diabetes?: Health be damned! Pour on the sugar. Diabetes Care 2014, 37, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high in fructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. 2014, 20, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Priebs, J.; Landmann, M.; Degen, C.; Engstler, A.J.; Jin, C.J.; Gärttner, S.; Spruss, A.; Huber, O.; Bergheim, I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 2015, 26, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H. Nutritional modulation of non-alcoholic fatty liver disease and insulin resistance. Nutrients 2015, 7, 9127–9138. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.A.; Gariani, K.; Oldani, G.; Delaune, V.; Morel, P.; Toso, C. Exercise-based interventions for nonalcoholic fatty liver disease: A meta-analysis and meta-regression. Clin. Gastroenterol. Hepatol. 2016, 14, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.-M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Sobrecases, H.; Lê, K.-A.; Bortolotti, M.; Schneiter, P.; Ith, M.; Kreis, R.; Boesch, C.; Tappy, L. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes Metab. 2010, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Lê, K.-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Samuel, V.T. The sweet path to metabolic demise: Fructose and lipid synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Nubret, E.; Sarfati, G.; Bergheim, I.; De Bandt, J.P. Citrulline and nonessential amino acids prevent fructose-induced nonalcoholic fatty liver disease in rats. J. Nutr. 2015, 145, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Freese, K.; Waligora-Dupriet, A.-J.; Nubret, E.; Butel, M.-J.; Bergheim, I.; De Bandt, J.P. Preventive effects of citrulline on Western diet-induced non-alcoholic fatty liver disease in rats. Br. J. Nutr. 2016, 116, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Cento, A.S.; Chiazza, F.; Collino, M.; Aragno, M. High-fructose intake as risk factor for neurodegeneration: Key role for carboxy methyllysine accumulation in mice hippocampal neurons. Neurobiol. Dis. 2016, 89, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Theytaz, F.; de Giorgi, S.; Hodson, L.; Stefanoni, N.; Rey, V.; Schneiter, P.; Giusti, V.; Tappy, L. Metabolic fate of fructose ingested with and without glucose in a mixed meal. Nutrients 2014, 6, 2632–2649. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Madlala, H.P.; Maarman, G.J.; Ojuka, E. Uric acid and transforming growth factor in fructose-induced production of reactive oxygen species in skeletal muscle. Nutr. Rev. 2016, 74, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Moran, G.; Estrada, A.; Pagliassotti, M.J. Fructose-induced stress signaling in the liver involves methylglyoxal. Nutr. Metab. (Lond.) 2013, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Lê, K.-A.; Faeh, D.; Stettler, R.; Debard, C.; Loizon, E.; Vidal, H.; Boesch, C.; Ravussin, E.; Tappy, L. Effects of four-week high-fructose diet on gene expression in skeletal muscle of healthy men. Diabetes Metab. 2008, 34, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Nigro, D.; Menotti, F.; Cento, A.S.; Serpe, L.; Chiazza, F.; dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic administration of saturated fats and fructose differently affect SREBP activity resulting in different modulation of Nrf2 and Nlrp3 inflammasome pathways in mice liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Foufelle, F.; Ferré, P. Unfolded protein response: Its role in physiology and physiopathology. Med. Sci. (Paris) 2007, 23, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Q.; Xu, C.-F.; Yu, C.-H.; Chen, W.-X.; Li, Y.-M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Zámbó, V.; Simon-Szabó, L.; Szelényi, P.; Kereszturi, E.; Bánhegyi, G.; Csala, M. Lipotoxicity in the liver. World J. Hepatol. 2013, 5, 550–557. [Google Scholar] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Galván-Peña, S.; O’Neill, L.A.J. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [PubMed]
- Brunt, E.M.; Tiniakos, D.G. Pathological features of NASH. Front. Biosci. 2005, 10, 1475–1484. [Google Scholar] [PubMed]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.-U. The role of hepatokines in metabolism. Nat. Rev. Endocrinol. 2013, 9, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Iroz, A.; Couty, J.-P.; Postic, C. Hepatokines: Unlocking the multi-organ network in metabolic diseases. Diabetologia 2015, 58, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Etienne-Mesmin, L.; Gewirtz, A.T. Microbiota-liver axis in hepatic disease. Hepatology 2014, 59, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-D.; Wang, Y.-H.; Chang, C.; Gershwin, M.E.; Lian, Z.-X. The intestinal microbiota and microenvironment in liver. Autoimmun. Rev. 2015, 14, 183–191. [Google Scholar] [CrossRef] [PubMed]
- De Bandt, J.-P.; Waligora-Dupriet, A.-J.; Butel, M.-J. Intestinal microbiota in inflammation and insulin resistance: Relevance to humans. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Sarfati, G.; Nubret, E.; Kapel, N.; Waligora-Dupriet, A.J.; Bergheim, I.; Cynober, L.; De-Bandt, J.P. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clin. Nutr. 2016, 35, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ritze, Y.; Bárdos, G.; Claus, A.; Ehrmann, V.; Bergheim, I.; Schwiertz, A.; Bischoff, S.C. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e80169. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, V.; di Gregorio, V.; Iebba, V.; Giusto, M.; Schippa, S.; Merli, M.; Thalheimer, U. Microbiota and the gut-liver axis: Bacterial translocation, inflammation and infection in cirrhosis. World J. Gastroenterol. 2014, 20, 16795–16810. [Google Scholar] [CrossRef] [PubMed]
- Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.M.; de Maria, S.; Cartenì, M.; Nardone, G. Gut–liver axis: The impact of gut microbiota on non alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [PubMed]
- Campos, V.C.; Tappy, L. Physiological handling of dietary fructose-containing sugars: Implications for health. Int. J. Obes. (Lond.) 2016, 40, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; van Thiel, D.H.; Lewis, P.J.S.; Ramadori, G. Insulin Production and Resistance in Different Models of Diet-Induced Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2017, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Vrána, A.; Fábry, P.; Kazdová, L. Effect of dietary fructose on fatty acid synthesis in adipose tissue and on triglyceride concentration in blood in the rat. Nutr. Metab. 1973, 15, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Masterjohn, C.; Park, Y.; Lee, J.; Noh, S.K.; Koo, S.I.; Bruno, R.S. Dietary fructose feeding increases adipose methylglyoxal accumulation in rats in association with low expression and activity of glyoxalase-2. Nutrients 2013, 5, 3311–3328. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wu, L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Heaney, A.P. Regulation of adipose differentiation by fructose and GluT5. Mol. Endocrinol. 2012, 26, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Sucrose, high-fructose corn syrup, and fructose, their metabolism and potential health effects: What do we really know? Adv. Nutr. 2013, 4, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Novell, J.M.; Ramió-Lluch, L.; Orozco, A.; Gómez-Foix, A.M.; Guinovart, J.J.; Rodríguez-Gil, J.E. Glucose and fructose have sugar-specific effects in both liver and skeletal muscle in vivo: A role for liver fructokinase. PLoS ONE 2014, 9, e109726. [Google Scholar] [CrossRef] [PubMed]
- Deldicque, L.; Cani, P.D.; Philp, A.; Raymackers, J.-M.; Meakin, P.J.; Ashford, M.L.J.; Delzenne, N.M.; Francaux, M.; Baar, K. The unfolded protein response is activated in skeletal muscle by high-fat feeding: Potential role in the downregulation of protein synthesis. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E695–E705. [Google Scholar] [CrossRef] [PubMed]
- Rieu, I.; Magne, H.; Savary-Auzeloux, I.; Averous, J.; Bos, C.; Peyron, M.A.; Combaret, L.; Dardevet, D. Reduction of low grade inflammation restores blunting of postprandial muscle anabolism and limits sarcopenia in old rats. J. Physiol. (Lond.) 2009, 587, 5483–5492. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; de Bandt, J.-P. Hepatic steatosis: A role for citrulline. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, D.; Ruiz, A.; Cabello-Verrugio, C.; Brandan, E.; Estrada, L.; Pizarro, M.; Solis, N.; Torres, J.; Barrera, F.; Arrese, M. Diet-induced nonalcoholic fatty liver disease is associated with sarcopenia and decreased serum insulin-like growth factor-1. Dig. Dis. Sci. 2016, 61, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Gatineau, E.; Savary-Auzeloux, I.; Migné, C.; Polakof, S.; Dardevet, D.; Mosoni, L. Chronic intake of sucrose accelerates sarcopenia in older male rats through alterations in insulin sensitivity and muscle protein synthesis. J. Nutr. 2015, 145, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Stephens, F.B.; Chee, C.; Wall, B.T.; Murton, A.J.; Shannon, C.E.; van Loon, L.J.C.; Tsintzas, K. Lipid-induced insulin resistance is associated with an impaired skeletal muscle protein synthetic response to amino acid ingestion in healthy young men. Diabetes 2015, 64, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. The effect of high-fat—High-fructose diet on skeletal muscle mitochondrial energetics in adult rats. Eur. J. Nutr. 2015, 54, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Sievenpiper, J.L.; de Souza, R.J.; Chiavaroli, L.; Ha, V.; Cozma, A.I.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Di Buono, M.; et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J. Nutr. 2012, 142, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Berglund, L.; McGahan, J.P.; Keim, N.L.; Havel, P.J. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr. Metab. (Lond.) 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hu, Y.; Huang, T.; Zhang, Y.; Li, Z.; Luo, C.; Luo, Y.; Yuan, H.; Hisatome, I.; Yamamoto, T.; et al. High uric acid directly inhibits insulin signalling and induces insulin resistance. Biochem. Biophys. Res. Commun. 2014, 447, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Serafini, M.; Colic Baric, I.; Hazen, S.L.; Klein, S. Effect of plasma uric acid on antioxidant capacity, oxidative stress, and insulin sensitivity in obese subjects. Diabetes 2014, 63, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.-Y.; Wu, H.-T.; Hung, H.-C.; Yang, Y.-C.; Wu, J.-S.; Chang, C.-J. Endoplasmic reticulum stress induces the expression of fetuin-A to develop insulin resistance. Endocrinology 2012, 153, 2974–2984. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Tomkiewicz, C.; Magne, L.; Barouki, R.; Garlatti, M. Endoplasmic reticulum stress induction of insulin-like growth factor-binding protein-1 involves ATF4. J. Biol. Chem. 2006, 281, 19124–19133. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, P.R.; Wagner, A.S.; Reddy, L.V.; Deutsch, D.D.; Leon, M.A.; Goustin, A.S.; Grunberger, G. Serum alpha 2-HS-glycoprotein is an inhibitor of the human insulin receptor at the tyrosine kinase level. Mol. Endocrinol. 1993, 7, 1445–1455. [Google Scholar] [PubMed]
- Crossey, P.A.; Jones, J.S.; Miell, J.P. Dysregulation of the insulin/IGF binding protein-1 axis in transgenic mice is associated with hyperinsulinemia and glucose intolerance. Diabetes 2000, 49, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, C.; Fang, Q.; Shao, M.; Zhang, Y.; Liu, Y.; Deng, Y.P.; Shan, B.; Liu, J.Q.; Li, H.T.; et al. Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J. Biol. Chem. 2014, 289, 29751–29765. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, Y.; Hu, Y.; Wang, G. The role of fibroblast growth factor 21 in the pathogenesis of non-alcoholic fatty liver disease and implications for therapy. Metabolism 2015, 64, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Ahmadian, M.; Yu, R.T.; Atkins, A.R.; Downes, M.; Evans, R.M. Nuclear receptors and metabolism: From feast to famine. Diabetologia 2014, 57, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.M.; Ahuja, S.P.; Marliss, E.B. Effects of intravenously administered fructose and glucose on splanchnic amino acid and carbohydrate metabolism in hypertriglyceridemic men. J. Clin. Investig. 1975, 56, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Hundal, H.S.; Darakhshan, F.; Kristiansen, S.; Blakemore, S.J.; Richter, E.A. GLUT5 expression and fructose transport in human skeletal muscle. Adv. Exp. Med. Biol. 1998, 441, 35–45. [Google Scholar] [PubMed]
- Laughlin, M.R. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 2014, 6, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Leverve, X. Rôle du foie dans le métabolisme des nutriments en nutrition artificielle. Nutr. Clin. Métabol. 1999, 13, 225–231. [Google Scholar] [CrossRef]
- Lazo, M.; Solga, S.F.; Horska, A.; Bonekamp, S.; Diehl, A.M.; Brancati, F.L.; Wagenknecht, L.E.; Pi-Sunyer, F.X.; Kahn, S.E.; Clark, J.M.; et al. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care 2010, 33, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Egli, L.; Lecoultre, V.; Theytaz, F.; Campos, V.; Hodson, L.; Schneiter, P.; Mittendorfer, B.; Patterson, B.W.; Fielding, B.A.; Gerber, P.A.; et al. Exercise prevents fructose-induced hypertriglyceridemia in healthy young subjects. Diabetes 2013, 62, 2259–2265. [Google Scholar] [CrossRef] [PubMed]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Schultze, F.C.; Wilting, J.; Mihm, S.; Raddatz, D.; Ramadori, G. Combination of alcohol and fructose exacerbates metabolic imbalance in terms of hepatic damage, dyslipidemia, and insulin resistance in rats. PLoS ONE 2014, 9, e104220. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Federico, A.; Dallio, M.; Scazzina, F. Mediterranean diet and nonalcoholic fatty liver disease: Molecular mechanisms of protection. Int. J. Food Sci. Nutr. 2017, 68, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Itsiopoulos, C.; Thodis, T.; Ward, G.; Trost, N.; Hofferberth, S.; O’Dea, K.; Desmond, P.V.; Johnson, N.A.; Wilson, A.M. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.M.; Johnson, N.A.; Burdon, C.A.; Cohn, J.S.; O’Connor, H.T.; George, J. Omega-3 supplementation and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 56, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Ramadori, G. How does bariatric surgery improve Type II Diabetes? The neglected importance of the liver in clearing glucose and insulin from the portal blood. J. Obes. Weight Loss Ther. 2015, 5, 280. [Google Scholar] [CrossRef]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, T.; Li, J.; Wang, S.; Qiu, F.; Yu, H.; Zhang, Y.; Wang, T. Effects of natural products on fructose-induced nonalcoholic fatty liver disease (NAFLD). Nutrients 2017, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Metformin protects against the development of fructose-induced steatosis in mice: Role of the intestinal barrier function. Lab. Investig. 2012, 92, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jegatheesan, P.; De Bandt, J. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. https://doi.org/10.3390/nu9030230
Jegatheesan P, De Bandt J. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients. 2017; 9(3):230. https://doi.org/10.3390/nu9030230
Chicago/Turabian StyleJegatheesan, Prasanthi, and Jean‐Pascal De Bandt. 2017. "Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism" Nutrients 9, no. 3: 230. https://doi.org/10.3390/nu9030230
APA StyleJegatheesan, P., & De Bandt, J. (2017). Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients, 9(3), 230. https://doi.org/10.3390/nu9030230