Cordyceps militaris Extract Protects Human Dermal Fibroblasts against Oxidative Stress-Induced Apoptosis and Premature Senescence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Cordyceps militaris Extract
2.3. Cell Culture
2.4. DPPH Radical Scavenging Activity
2.5. Cell Viability Analysis
2.6. Intracellular ROS Scavenging Activity
2.7. Measurement of DNA Condensation
2.8. Apoptosis Analysis
2.9. Senescence-Associated β-Galactosidase Staining
2.10. Statistical Analysis
3. Results
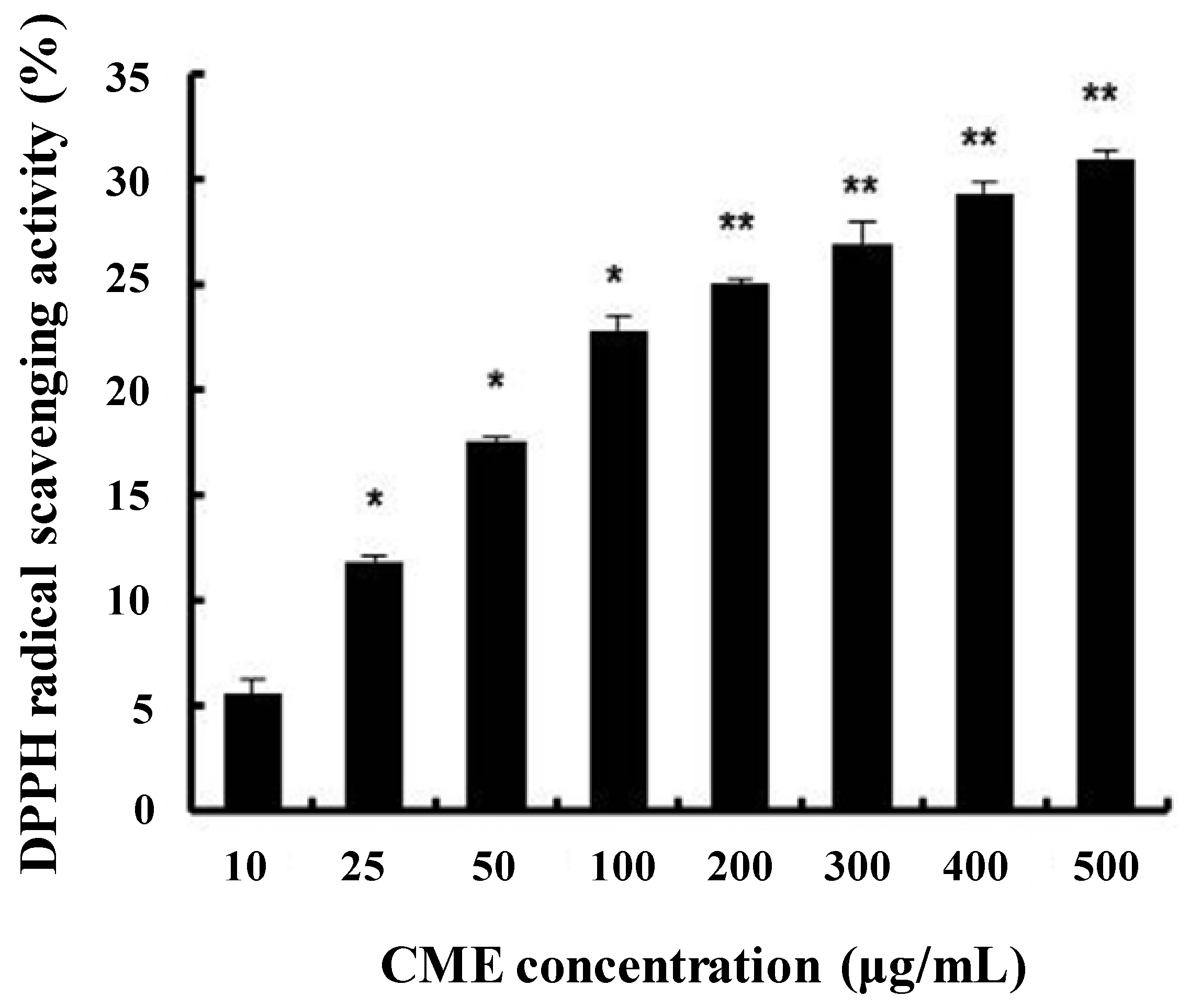
3.1. DPPH Radical Scavenging Activity of CME
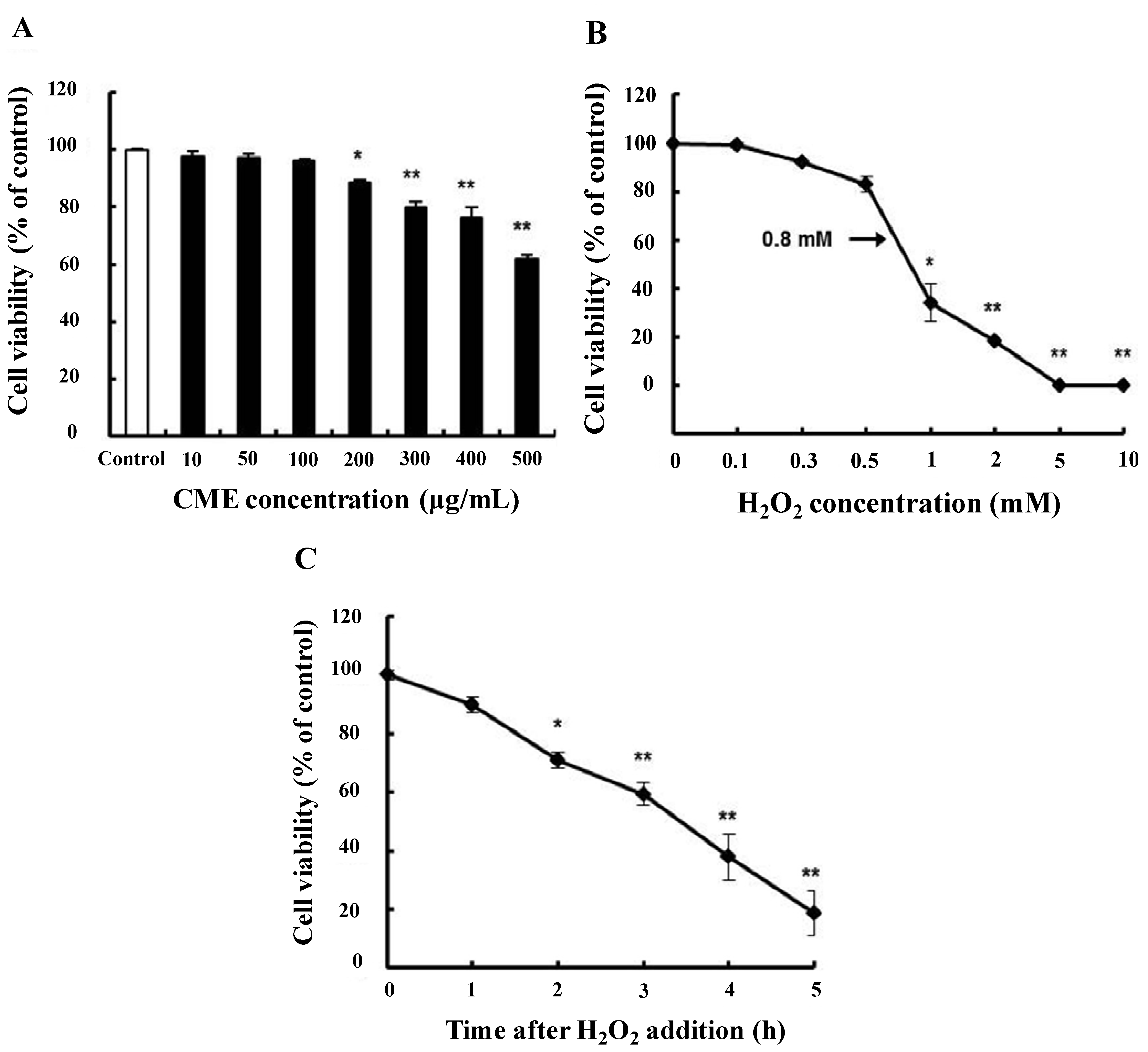
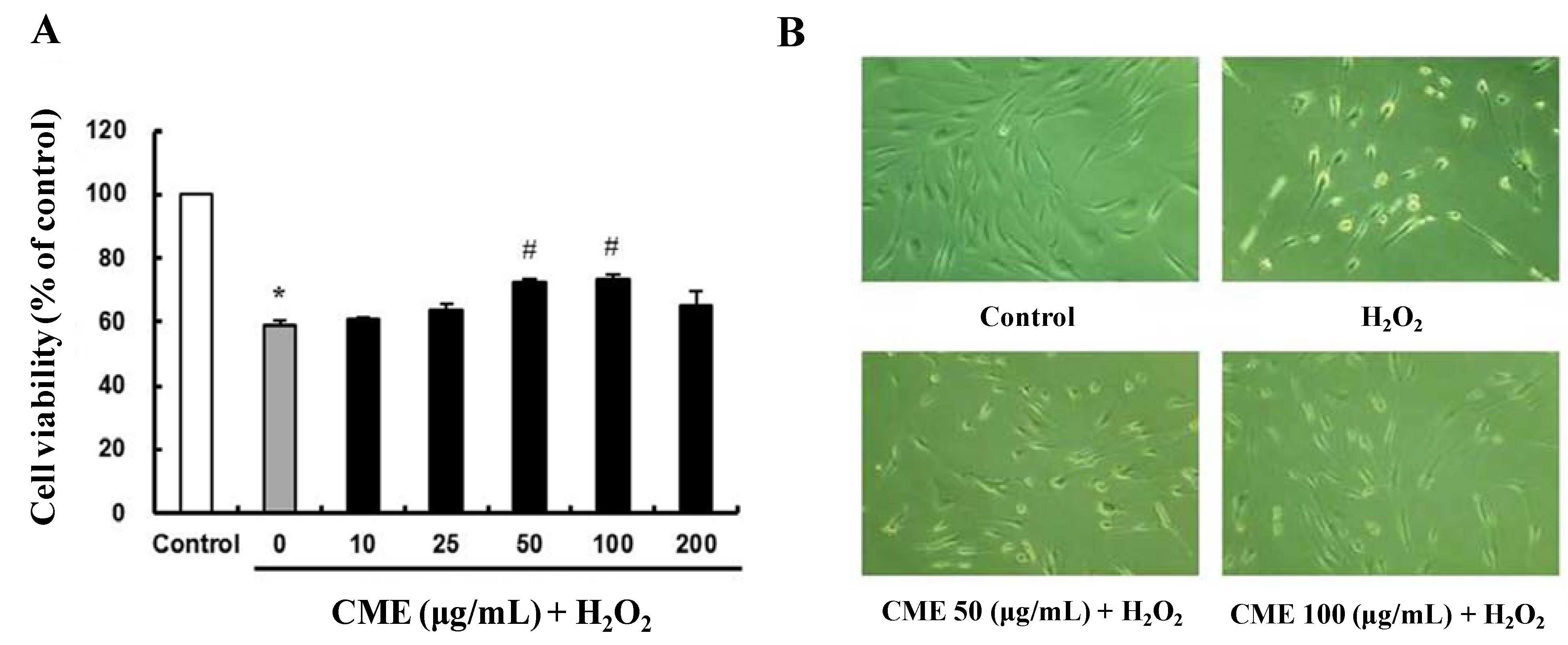
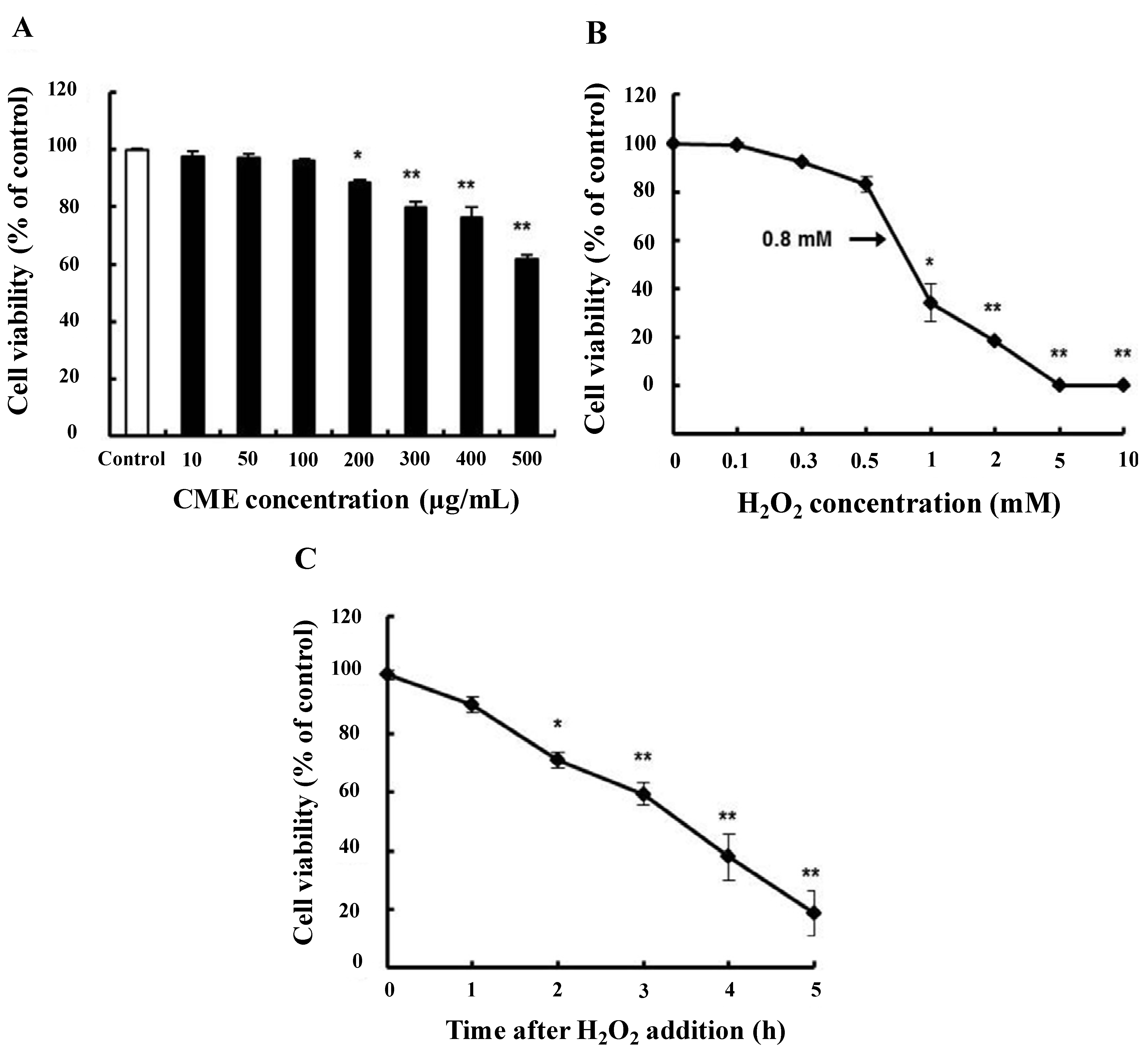
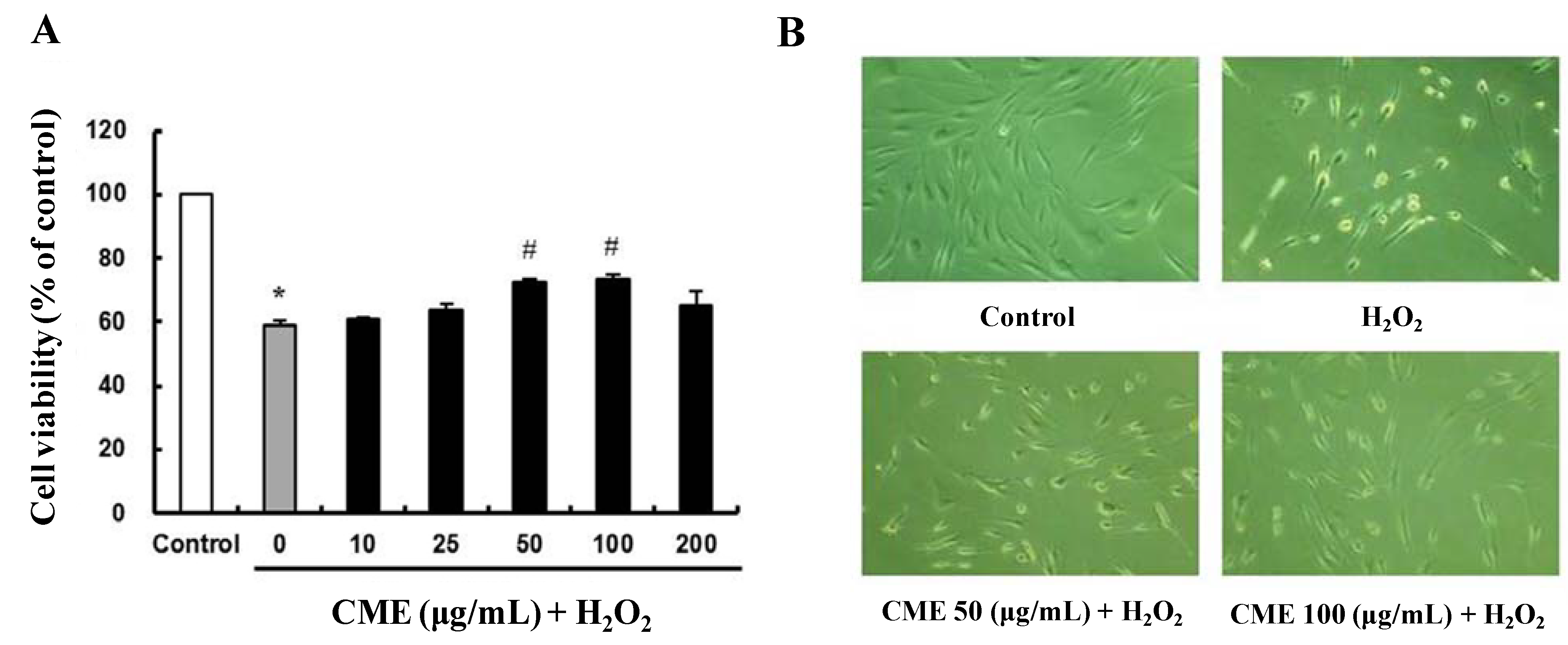
3.2. Cytoprotective Effect of CME against Hydrogen Peroxide-Induced Oxidative Stress in HDFs
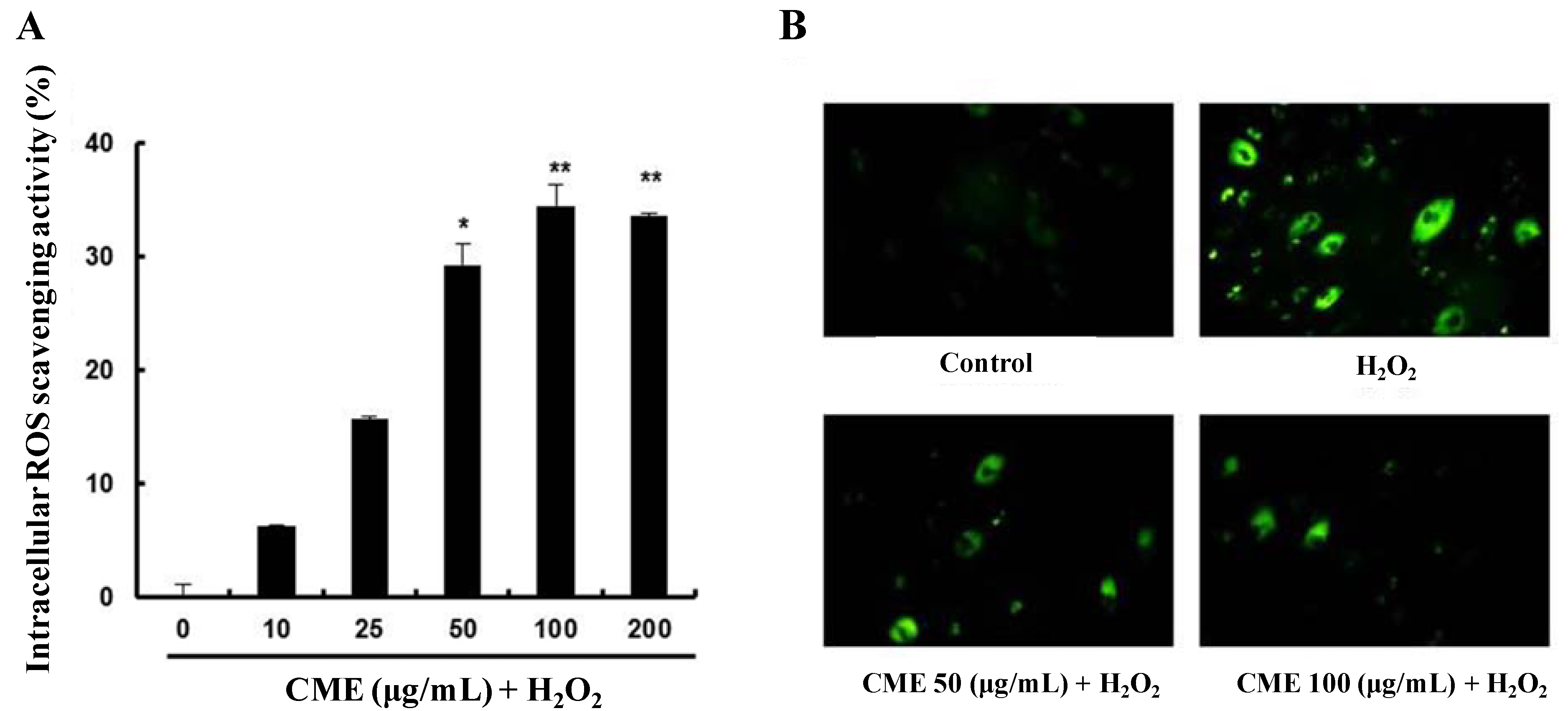
3.3. CME Inhibited Hydrogen Peroxide-Induced ROS Generation in HDFs

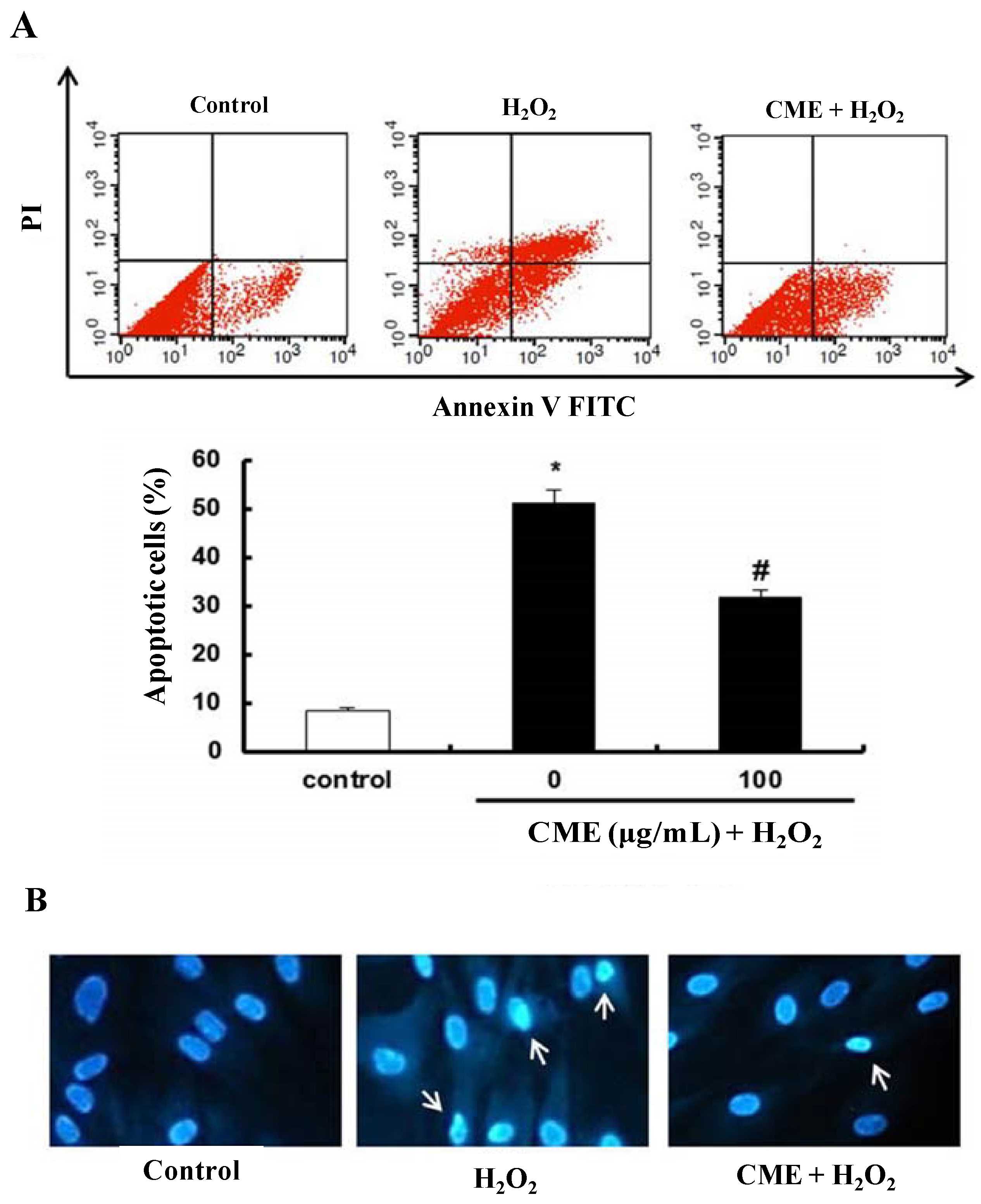
3.4. Effect of CME on Hydrogen Peroxide-Induced Apoptotic Cell Death in HDFs
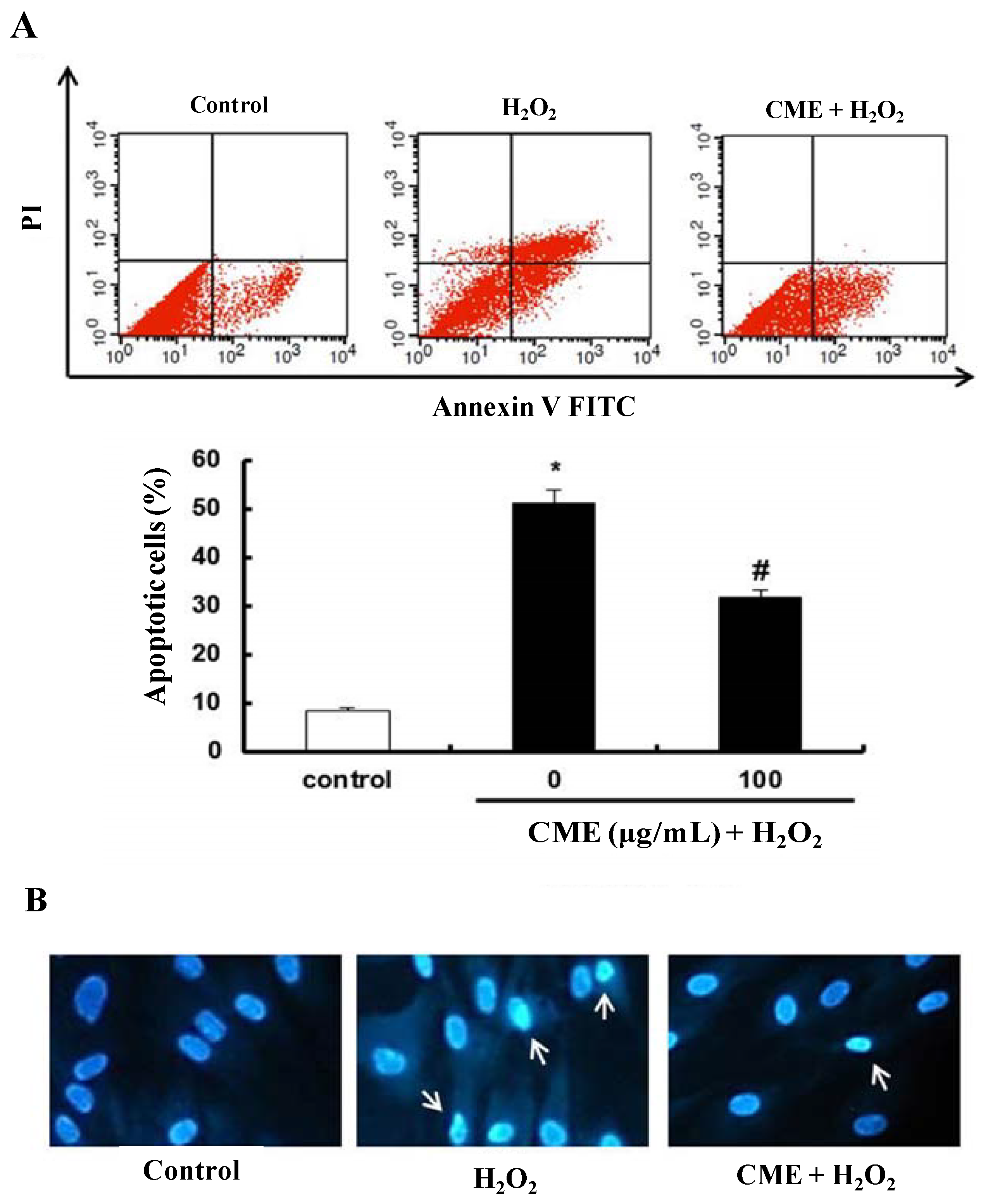
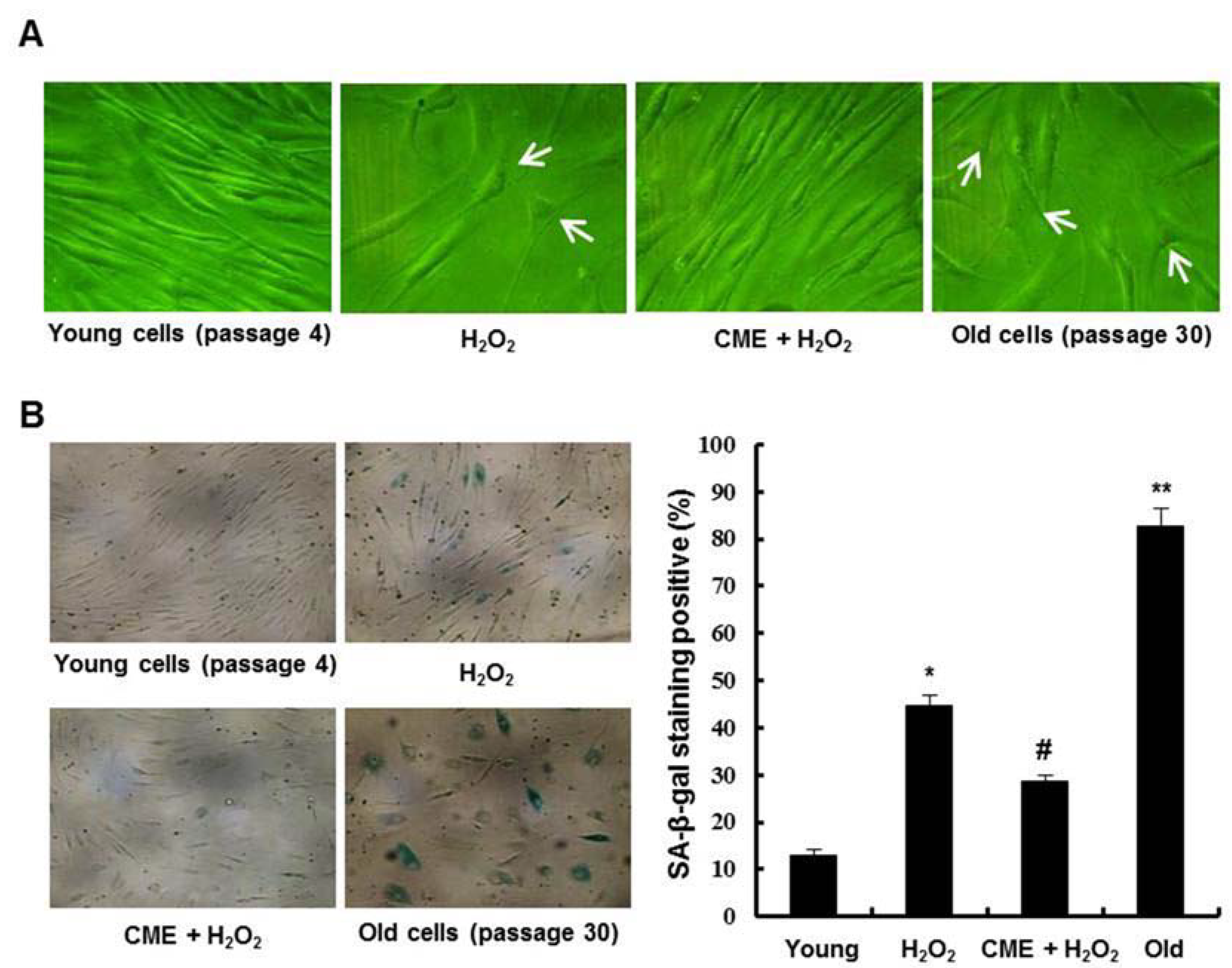
3.5. Effect of CME on Hydrogen Peroxide-Induced Premature Senescence in HDFs
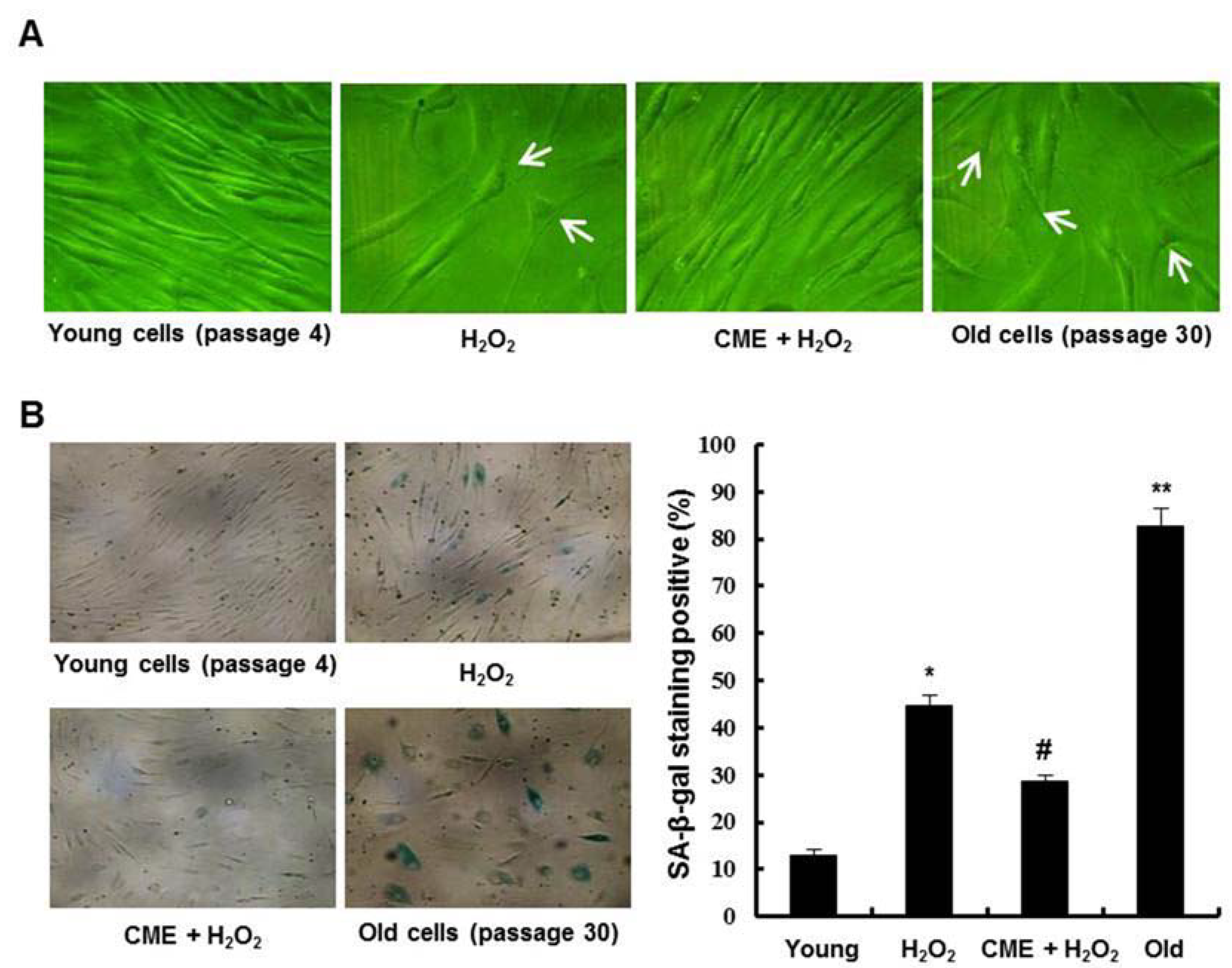
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El-Domyati, M.; Attia, S.; Saleh, F.; Brown, D.; Birk, D.E.; Gasparro, F.; Ahmad, H.; Uitto, J. Intrinsic aging vs. photoaging: A comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp. Dermatol. 2002, 11, 398–405. [Google Scholar]
- Bhawan, J.; Andersen, W.; Lee, J.; Labadie, R.; Solares, G. Photoaging vs. intrinsic aging: A morphologic assessment of facial skin. J. Cutan. Pathol. 1995, 22, 154–159. [Google Scholar]
- Uitto, J. The role of elastin and collagen in cutaneous aging: Intrinsic aging vs. photoexposure. J. Drugs Dermatol. 2008, 7, s12–s16. [Google Scholar]
- Scharffetter-Kochanek, K.; Wlaschek, M.; Brenneisen, P.; Schauen, M.; Blaudschun, R.; Wenk, J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol. Chem 1997, 378, 1247–1257. [Google Scholar]
- Jacobson, M.D. Reactive oxygen species and programmed cell death. Trends Biochem. Sci. 1996, 21, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Free radical theory of aging. Mutat. Res. 1992, 275, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Burton, M.; Chen, Q.M.; Gonos, E.S.; Frippiat, C.; Mazarati, J.B.; Eliaers, F.; Remacle, J.; Toussaint, O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic. Biol. Med. 2000, 28, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Burova, E.; Borodkina, A.; Shatrova, A.; Nikolsky, N. Sublethal oxidative stress induces the premature senescence of human mesenchymal stem cells derived from endometrium. Oxid. Med. Cell. Longev. 2013, 2013, 474931. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar]
- Chaturvedi, V.; Sitailo, L.A.; Qin, J.Z.; Bodner, B.; Denning, M.F.; Curry, J.; Zhang, W.G.; Brash, D.; Nickoloff, B.J. Knockdown of p53 levels in human keratinocytes accelerates Mcl-1 and Bcl-x(l) reduction thereby enhancing UV-light induced apoptosis. Oncogene 2005, 24, 5299–5312. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Qin, J.Z.; Stennett, L.; Choubey, D.; Nickoloff, B.J. Resistance to UV-induced apoptosis in human keratinocytes during accelerated senescence is associated with functional inactivation of p53. J. Cell. Physiol. 2004, 198, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Oresajo, C.; Hayward, J. Ultraviolet radiation and skin aging: Roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation—A review. Int. J. Cosmet. Sci. 2005, 27, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gong, Z.; Su, Y.; Lin, J.; Tang, K. Cordyceps fungi: Natural products. J. Pharm. Pharmacol. 2009, 61, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Wu, G.H.; Huang, Z.L. Structural analysis and antioxidant activities of polysaccharides from cultured Cordyceps militaris. Int. J. Biol. Macromol. 2013, 58, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Won, S.Y.; Park, E.H. Anti-inflammatory and related pharmacological activities of cultured mycelia and fruiting bodies of Cordyceps militaris. J. Ethnopharmacol. 2005, 96, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Huang, Y.D.; Bian, Y.; Wong, J.H.; Ng, T.B.; Wang, H.X. Hypoglycemic activity of the fungi Cordyceps militaris, Cordyceps sinensis, Tricholoma mongolicum, and Omphalia lapidescens in streptozotocin-induced diabetic rats. Appl. Microbiol. Biot. 2006, 72, 1152–1156. [Google Scholar] [CrossRef]
- Yu, R.M.; Yang, W.; Song, L.Y.; Yan, C.Y.; Zhang, Z.; Zhao, Y. Structural characterization and antioxidant activity of a polysaccharide from the fruiting bodies of cultured Cordyceps militaris. Carbohyd. Polym. 2007, 70, 430–436. [Google Scholar] [CrossRef]
- Yan, H.; Zhu, D.J.; Xu, D.B.; Wu, J.; Bian, X.Y. A study on Cordyceps militaris polysaccharide purification, composition and activity analysis. Afr. J. Biotechnol. 2008, 7, 4004–4009. [Google Scholar]
- Weng, Y.F.; Lu, J.; Xiang, L.; Matsuura, A.; Zhang, Y.; Huang, Q.M.; Qi, J.H. Ganodermasides C and D, two new anti-aging ergosterols from spores of the medicinal mushroom Ganoderma lucidum. Biosci. Biotechnol. Biochem. 2011, 75, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.P.; Wang, C.M.; Tsui, T.Y.; Wai, M.S.; Tang, H.C.; Wong, Y.W.; Lam, L.H.; Hui, L.K.; Yew, D.T. Extract of white button mushroom affects skin healing and angiogenesis. Microsc. Res. Tech. 2012, 75, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Koleva, I.I.; van Beek, T.A.; Linssen, J.P.; de Groot, A.; Evstatieva, L.N. Screening of plant extracts for antioxidant activity: A comparative study on three testing methods. Phytochem. Anal. 2002, 13, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Gary, R.K.; Kindell, S.M. Quantitative assay of senescence-associated beta-galactosidase activity in mammalian cell extracts. Anal. Biochem. 2005, 343, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Toledo, R.T. Major flavonoids in grape seeds and skins: Antioxidant capacity of catechin, epicatechin, and gallic acid. J. Agric. Food Chem. 2004, 52, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.P.; Lin, S.Q.; Jiang, Y.M.; Ashraf, M. Variation in contents of total phenolics and flavonoids and antioxidant activities in the leaves of 11 Eriobotrya species. Plant Food Hum. Nutr. 2008, 63, 200–204. [Google Scholar] [CrossRef]
- Lin, R.S.; Liu, H.H.; Wu, S.Q.; Pang, L.F.; Jia, M.S.; Fan, K.M.; Jia, S.H.; Jia, L. Production and in vitro antioxidant activity of exopolysaccharide by a mutant, Cordyceps militaris SU5-08. Int. J. Biol. Macromol. 2012, 51, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Duan, J.; Zhang, Z.; Tong, T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int. J Biochem. Cell. Biol. 2005, 37, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.E.; Meneghini, R. Action of hydrogen peroxide on human fibroblast in culture. Photochem. Photobiol. 1979, 30, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, M.K.; Joshi, P.N.; Nair, J.S.; Ramaswamy, N.K.; Iyer, R.K.; Biswal, B.; Biswal, U.C. UV-B exposure enhances senescence of wheat leaves: Modulation by photosynthetically active radiation. Radiat. Environ. Biophys. 2006, 45, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Jee, H.J.; Kim, H.J.; Kim, A.J.; Bae, Y.S.; Bae, S.S.; Yun, J. UV light induces premature senescence in Akt1-null mouse embryonic fibroblasts by increasing intracellular levels of ROS. Biochem. Biophys. Res. Commun. 2009, 383, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Emri, G.; Horkay, I.; Remenyik, E. The role of free radicals in the UV-induced skin damage. Photo-aging. Orv. Hetil. 2006, 147, 731–735. (in Hungarian). [Google Scholar]
- Ngawhirunpat, T.; Opanasopi, P.; Sukma, M.; Sittisombut, C.; Kat, A.; Adachi, I. Antioxidant, free radical-scavenging activity and cytotoxicity of different solvent extracts and their phenolic constituents from the fruit hull of mangosteen (Garcinia mangostana). Pharm. Biol. 2010, 48, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kutuk, O.; Adli, M.; Poli, G.; Basaga, H. Resveratrol protects against 4-HNE induced oxidative stress and apoptosis in Swiss 3T3 fibroblasts. Biofactors 2004, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Han, D.W.; Kim, H.H.; Son, H.J.; Baek, H.S.; Lee, K.Y.; Hyon, S.H.; Park, J.C. Protection of human fibroblasts from reactive oxygen species by green tea polyphenolic compounds. Key Eng. Mater. 2005, 288–289, 665–668. [Google Scholar]
- Zhan, Y.; Dong, C.H.; Yao, Y.J. Antioxidant activities of aqueous extract from cultivated fruit-bodies of Cordyceps militaris (L.) link in vitro. J. Integr. Plant Biol. 2006, 48, 1365–1370. [Google Scholar]
- Gurjala, A.N.; Liu, W.R.; Mogford, J.E.; Procaccini, P.S.A.; Mustoe, T.A. Age-dependent response of primary human dermal fibroblasts to oxidative stress: Cell survival, pro-survival kinases, and entrance into cellular senescence. Wound Repair Regen. 2005, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Ma, L.J.; Yao, J.J.; Fang, Y.; Mei, Y.A.; Wei, S.M. Protective effect of oat bran extracts on human dermal fibroblast injury induced by hydrogen peroxide. J. Zhejiang Univ. Sci. B 2013, 14, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.; Esposti, M.D.; Gilmore, A.P. Bcl-2 proteins and mitochondria—Specificity in membrane targeting for death. Biochim. Biophys. Acta 2011, 1813, 532–539. [Google Scholar]
- Adams, J.M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol. 2007, 19, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Marsden, V.S.; O’Connor, L.; O’Reilly, L.A.; Silke, J.; Metcalf, D.; Ekert, P.G.; Huang, D.C.; Cecconi, F.; Kuida, K.; Tomaselli, K.J.; et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 2002, 419, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Harte, M.T.; Kluck, R.M.; Wolf, B.B.; Casiano, C.A.; Newmeyer, D.D.; Wang, H.G.; Reed, J.C.; Nicholson, D.W.; Alnemri, E.S.; et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2,-3,-6,-7,-8,and -10 in a caspase-9-dependent manner. J. Cell Biol. 1999, 144, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Testori, A.; Acosta, M.; Campisi, J. Replicative senescence, aging and growth-regulatory transcription factors. Biol. Signals 1996, 5, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Comings, D.E.; Okada, T.A. Electron microscopy of human fibroblasts in tissue culture during logarithmic and confluent stages of growth. Exp.Cell Res. 1970, 61, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Bassaneze, V.; Miyakawa, A.A.; Krieger, J.E. A quantitative chemiluminescent method for studying replicative and stress-induced premature senescence in cell cultures. Anal. Biochem. 2008, 372, 198–203. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Park, J.M.; Lee, J.S.; Lee, K.R.; Ha, S.-J.; Hong, E.K. Cordyceps militaris Extract Protects Human Dermal Fibroblasts against Oxidative Stress-Induced Apoptosis and Premature Senescence. Nutrients 2014, 6, 3711-3726. https://doi.org/10.3390/nu6093711
Park JM, Lee JS, Lee KR, Ha S-J, Hong EK. Cordyceps militaris Extract Protects Human Dermal Fibroblasts against Oxidative Stress-Induced Apoptosis and Premature Senescence. Nutrients. 2014; 6(9):3711-3726. https://doi.org/10.3390/nu6093711
Chicago/Turabian StylePark, Jun Myoung, Jong Seok Lee, Ki Rim Lee, Suk-Jin Ha, and Eock Kee Hong. 2014. "Cordyceps militaris Extract Protects Human Dermal Fibroblasts against Oxidative Stress-Induced Apoptosis and Premature Senescence" Nutrients 6, no. 9: 3711-3726. https://doi.org/10.3390/nu6093711
APA StylePark, J. M., Lee, J. S., Lee, K. R., Ha, S.-J., & Hong, E. K. (2014). Cordyceps militaris Extract Protects Human Dermal Fibroblasts against Oxidative Stress-Induced Apoptosis and Premature Senescence. Nutrients, 6(9), 3711-3726. https://doi.org/10.3390/nu6093711