Metabolic Interactions between Vitamin A and Conjugated Linoleic Acid


Abstract
:1. Introduction
2. Vitamin A Metabolism and Functions
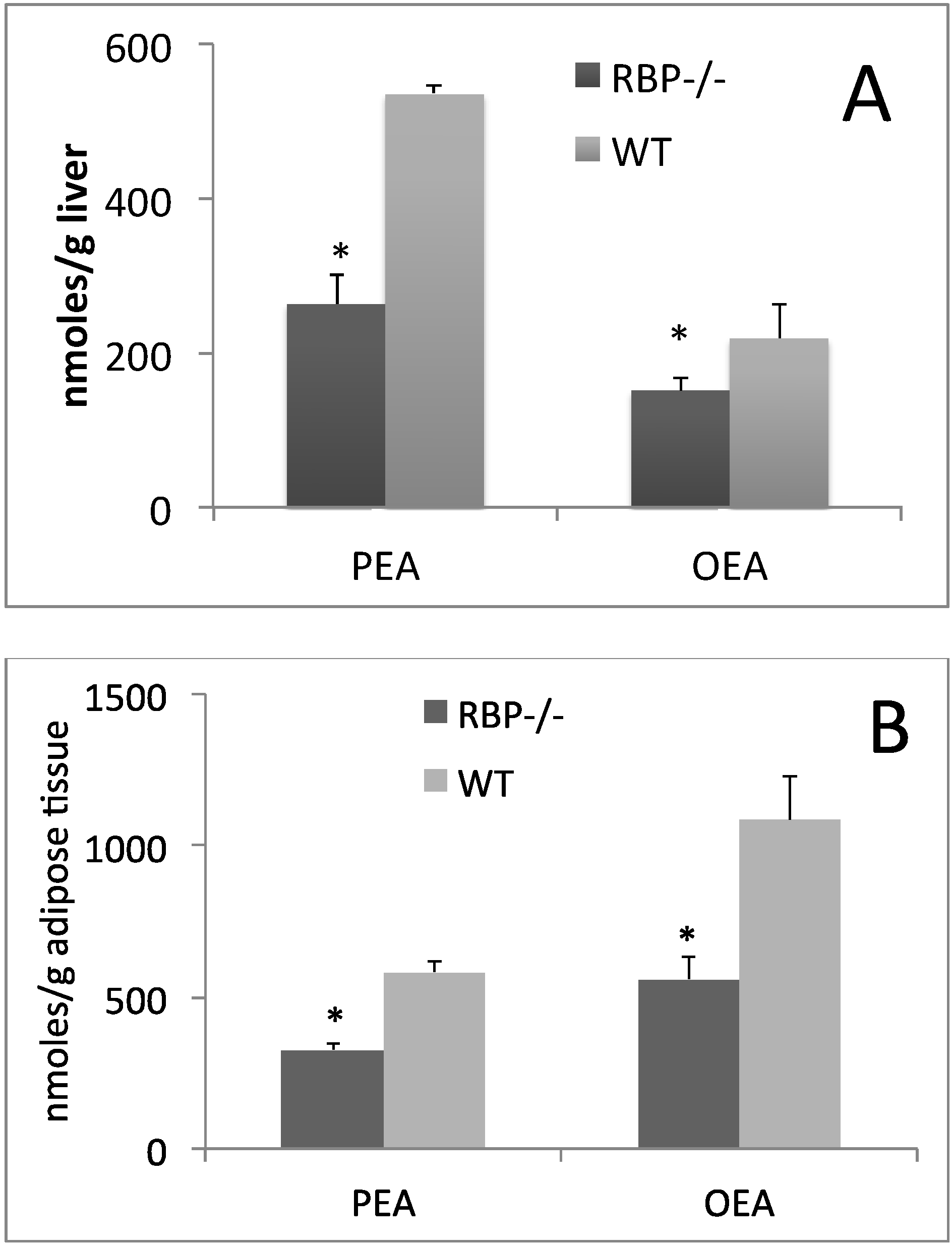
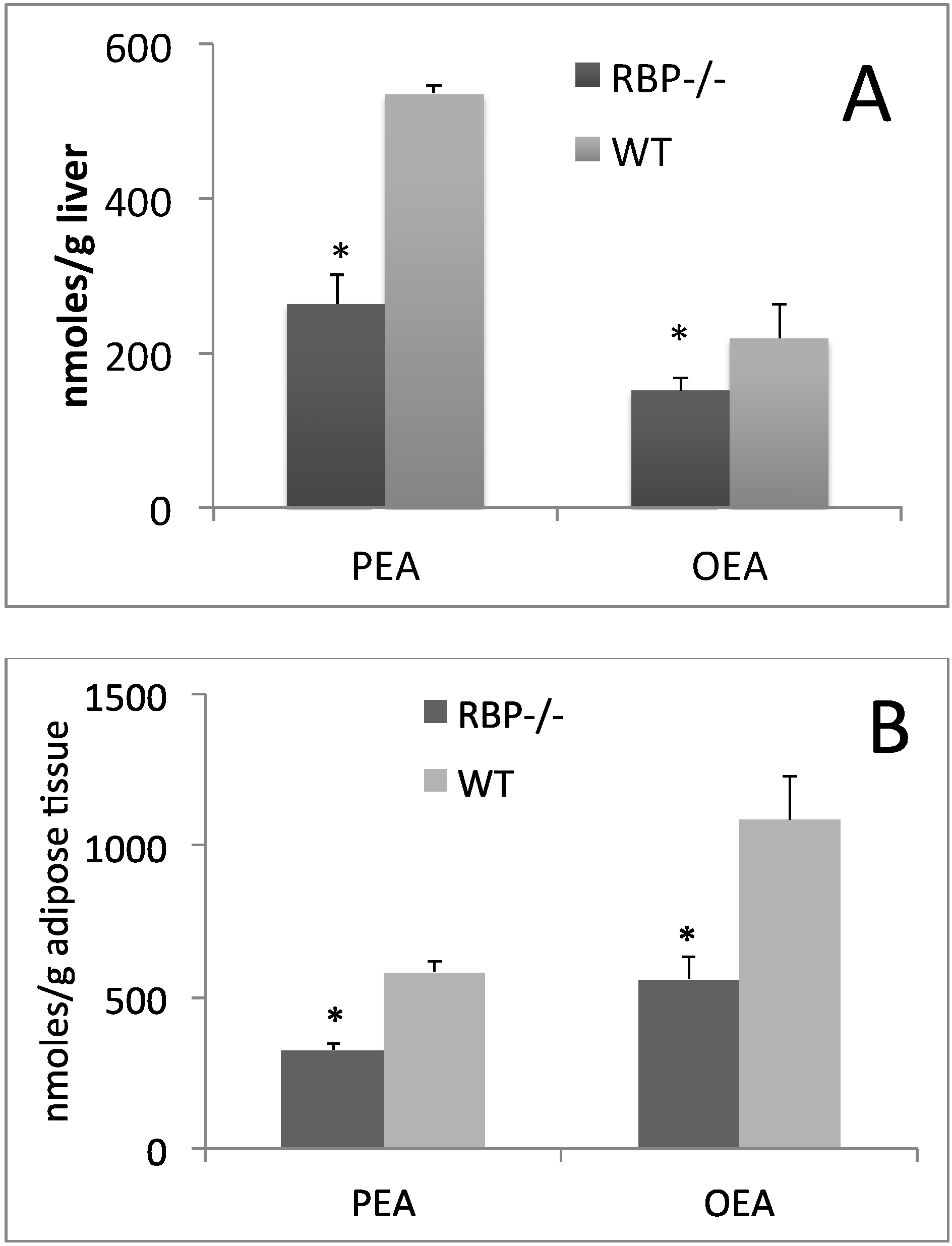
3. Influence of Dietary CLA on Retinol and RBP Tissue Levels
4. Is PPAR-α the Key Regulator of CLA and Retinoid Metabolic Routes?

{kind=link}
| Treatment | Retinol | Retinyl Esters |
|---|---|---|
| μg/g of Liver | ||
| CTRL | 4.69 ± 0.73 a | 273.69 ± 67.37 a |
| CLA | 6.68 ± 2.03 a | 470.64 ± 46.06 b |
| WY-14,643 | 1.41 ± 0.45 b | 75.74 ± 14.23 c |
5. Conclusions
Conflicts of Interest
References
- McIntosh, F.M.; Shingfield, K.J.; Devillard, E.; Russell, W.R.; Wallace, R.J. Mechanism of conjugated linoleic acid and vaccenic acid formation in human faecal suspensions and pure cultures of intestinal bacteria. Microbiology 2009, 155, 285–294. [Google Scholar] [CrossRef]
- Griinari, J.M.; Cori, B.A.; Lacy, S.H.; Chouinard, P.Y.; Nurmela, K.V.V.; Bauman, D.E. Conjugated linoleic acid is synthesized endogenously in lactating dairy cows by δ(9)-desaturase. J. Nutr. 2000, 130, 2285–2291. [Google Scholar]
- Ritzenthaler, K.L.; McGuire, M.K.; Falen, R.; Shultz, T.D.; Dasgupta, N.; McGuire, M.A. Estimation of conjugated linoleic acid intake by written dietary assessment methodologies underestimates actual intake evaluated by food duplicate methodology. J. Nutr. 2001, 131, 1548–1554. [Google Scholar]
- Pariza, M.W.; Ha, Y.L. Newly recognized anticarcinogenic fatty acids. Basic Life Sci. 1990, 52, 167–170. [Google Scholar]
- Ma, D.W.L.; Wierzbicki, A.A.; Field, C.J.; Clandinin, M.T. Preparation of conjugated linoleic acid from safflower oil. J. Am. Oil Chem. Soc. 1999, 76, 729–730. [Google Scholar] [CrossRef]
- Belury, M.A. Dietary conjugated linoleic acid in health: Physiological effects and mechanisms of action. Ann. Rev. Nutr. 2002, 22, 505–531. [Google Scholar] [CrossRef]
- Kennedy, A.; Martinez, K.; Schmidt, S.; Mandrup, S.; LaPoint, K.; McIntosh, M. Antiobesity mechanisms of action of conjugated linoleic acid. J. Nutr. Biochem. 2010, 21, 171–179. [Google Scholar] [CrossRef]
- Jiang, S.; Chen, H.; Wang, Z.; Riethoven, J.J.; Xia, Y.; Miner, J.; Fromm, M. Activated AMPK and prostaglandins are involved in the response to conjugated linoleic acid and are sufficient to cause lipid reductions in adipocytes. J. Nutr. Biochem. 2011, 22, 656–664. [Google Scholar] [CrossRef]
- Moya-Camarena, S.Y.; Vanden Heuvel, J.P.; Blanchard, S.G.; Leesnitzer, L.A.; Belury, M.A. Conjugated linoleic acid is a potent naturally occurring ligand and activator of PPARα. J. Lipid Res. 1999, 40, 1426–1433. [Google Scholar]
- Peters, J.M.; Park, Y.; Gonzalez, F.J.; Pariza, M.W. Influence of conjugated linoleic acid on body composition and target gene expression in peroxisome proliferator-activated receptor α-null mice. Biochim. Biophys. Acta 2001, 1533, 233–242. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the X-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor α-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar]
- Grimaldi, P.A. Metabolic and nonmetabolic regulatory functions of peroxisome proliferator-activated receptor β. Curr. Opin. Lipidol. 2010, 21, 186–191. [Google Scholar] [CrossRef]
- Sanderson, L.M.; Degenhardt, T.; Koppen, A.; Kalkhoven, E.; Desvergne, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor β/δ (PPAR β/δ) but not PPARα serves as a plasma free fatty acid sensor in liver. Mol. Cell. Biol. 2009, 29, 6257–6267. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. Ppargamma: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptorsα and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar]
- Banni, S.; Angioni, E.; Casu, V.; Melis, M.P.; Scrugli, S.; Carta, G.; Corongiu, F.P.; Ip, C. An increase in vitamin a status by the feeding of conjugated linoleic acid. Nutr. Cancer 1999, 33, 53–57. [Google Scholar] [CrossRef]
- Goodman, D.S. Vitamin A and retinoids in health and disease. N. Engl. J. Med. 1984, 310, 1023–1031. [Google Scholar] [CrossRef]
- Napoli, J.L. Biochemical pathways of retinoid transport, metabolism, and signal transduction. Clin. Immunol. Immunopathol. 1996, 80, S52–S62. [Google Scholar] [CrossRef]
- Saari, J.C. Retinoids in Mammalian Vision. In Retinoids; Nau, H., Blaner, W.S., Eds.; Springer Verlag Publishing: Heidelberg, Germany, 1999; Volume 139, pp. 563–588. [Google Scholar]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Kurokawa, R.; Soderstrom, M.; Horlein, A.; Halachmi, S.; Brown, M.; Rosenfeld, M.G.; Glass, C.K. Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature 1995, 377, 451–454. [Google Scholar] [CrossRef]
- Leblanc, B.P.; Stunnenberg, H.G. 9-cis retinoic acid signaling: Changing partners causes some excitement. Genes Dev. 1995, 9, 1811–1816. [Google Scholar] [CrossRef]
- Chen, J.D.; Evans, R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar] [CrossRef]
- Pfahl, M.; Chytil, F. Regulation of metabolism by retinoic acid and its nuclear receptors. Ann. Rev. Nutr. 1996, 16, 257–283. [Google Scholar] [CrossRef]
- Kastner, P.; Mark, M.; Chambon, P. Nonsteroid nuclear receptors: What are genetic studies telling us about their role in real life? Cell 1995, 83, 859–869. [Google Scholar] [CrossRef]
- Sporn, M.B.; Roberts, A.B.; Goodman, D.S. The Retinoids: Biology, Chemistry, and Medicine, 2nd ed.; Raven Press: New York, NY, USA, 1994. [Google Scholar]
- Vogel, S.; Gamble, M.V.; Blaner, W.S. Biosynthesis, Absorption, Metabolism and Transport of Retinoids. In Retinoids—The Biochemical and Molecular Basis of Vitamin A and Retinoid Action; Nau, H., Blaner, W.S., Eds.; Springer Verlag Publishing: Heidelberg, Germany, 1999; Volume 139, pp. 31–95. [Google Scholar]
- Goldberg, I.J. Lipoprotein lipase and lipolysis: Central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 1996, 37, 693–707. [Google Scholar]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar]
- Blaner, W.S.; Olson, J.A. Retinol and Retinoic Acid Metabolism. In The Retinoids, Biology, Chemistry and Medicine; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 229–256. [Google Scholar]
- Goodman, D.S.; Huang, H.S.; Shiratori, T. Tissue distribution of newly absorbed vitamin A in the rat. J. Lipid Res. 1965, 6, 390–396. [Google Scholar]
- Quadro, L.; Hamberger, L.; Colantuoni, V.; Gottesman, M.E.; Blaner, W.S. Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mol. Aspects Med. 2003, 24, 421–430. [Google Scholar] [CrossRef]
- Soprano, D.R.; Blaner, W.S. Plasma Retinol-Binding Protein. In The Retinoids, Biology, Chemistry and Medicine; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 257–282. [Google Scholar]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar]
- Biesalski, H.K.; Frank, J.; Beck, S.C.; Heinrich, F.; Illek, B.; Reifen, R.; Gollnick, H.; Seeliger, M.W.; Wissinger, B.; Zrenner, E. Biochemical but not clinical vitamin a deficiency results from mutations in the gene for retinol-binding protein. Am. J. Clin. Nutr. 1999, 69, 931–936. [Google Scholar]
- Dugan, M.; Rolland, D.C.; Best, D.R. The effects of feeding conjugated linoleic acid on pig liver vitamin A and retinol binding protein mRNA. Can. J. Anim. Sci. 2002, 82, 461–463. [Google Scholar] [CrossRef]
- Ortiz, B.; Wassef, L.; Shabrova, E.; Cordeddu, L.; Banni, S.; Quadro, L. Hepatic retinol secretion and storage are altered by dietary CLA: Common and distinct actions of CLA c9, t11 and t10, c12 isomers. J. Lipid Res. 2009, 50, 2278–2289. [Google Scholar] [CrossRef]
- Andreola, F.; Hayhurst, G.P.; Luo, G.; Ferguson, S.S.; Gonzalez, F.J.; Goldstein, J.A.; de Luca, L.M. Mouse liver CYP2C39 is a novel retinoic acid 4-hydroxylase. Its down-regulation offers a molecular basis for liver retinoid accumulation and fibrosis in aryl hydrocarbon receptor-null mice. J. Biol. Chem. 2004, 279, 3434–3438. [Google Scholar]
- White, J.A.; Guo, Y.D.; Baetz, K.; Beckett-Jones, B.; Bonasoro, J.; Hsu, K.E.; Dilworth, F.J.; Jones, G.; Petkovich, M. Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J. Biol. Chem. 1996, 271, 29922–29927. [Google Scholar] [CrossRef]
- Johnson, E.F.; Palmer, C.N.; Griffin, K.J.; Hsu, M.H. Role of the peroxisome proliferator-activated receptor in cytochrome P450 4A gene regulation. FASEB J. 1996, 10, 1241–1248. [Google Scholar]
- Belury, M.A.; Moya-Camarena, S.Y.; Liu, K.L.; Vanden Heuvel, J.P. Dietary conjugated linoleic acid induces peroxisome-specific enzyme accumulation and ornithine decarboxylase activity in mouse liver. J. Nutr. Biochem. 1997, 8, 579–584. [Google Scholar] [CrossRef]
- Clement, L.; Poirier, H.; Niot, I.; Bocher, V.; Guerre-Millo, M.; Krief, S.; Staels, B.; Besnard, P. Dietary trans-10, cis-12 conjugated linoleic acid induces hyperinsulinemia and fatty liver in the mouse. J. Lipid Res. 2002, 43, 1400–1409. [Google Scholar] [CrossRef]
- Seo, J.A.; Kim, N.H.; Park, S.Y.; Kim, H.Y.; Ryu, O.H.; Lee, K.W.; Lee, J.; Kim, D.L.; Choi, K.M.; Baik, S.H.; et al. Serum retinol-binding protein 4 levels are elevated in non-alcoholic fatty liver disease. Clin. Endocrinol. 2008, 68, 555–560. [Google Scholar] [CrossRef]
- Giordano, E.; Banni, S.; Quadro, L. A single dose of c9, t11 or t10, c12 conjugated linoleic acid isomers perturbs vitamin A metabolism in mice. Nutr. Res. 2011, 31, 855–862. [Google Scholar] [CrossRef]
- Keller, H. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA 1993, 90, 2160–2164. [Google Scholar] [CrossRef]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: Genomic and nongenomic effects. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef]
- Lengqvist, J.; Mata de Urquiza, A.; Bergman, A.C.; Willson, T.M.; Sjovall, J.; Perlmann, T.; Griffiths, W.J. Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor α ligand-binding domain. Mol. Cell. Proteomics 2004, 3, 692–703. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010, 2010, 1–20. [Google Scholar] [CrossRef]
- Iannone, A.; Petroni, A.; Murru, E.; Cordeddu, L.; Carta, G.; Melis, M.P.; Bergamini, S.; Casa, L.D.; Cappiello, L.; Carissimi, R.; et al. Impairment of 8-iso-PGF (2α) isoprostane metabolism by dietary conjugated linoleic acid (CLA). Prostaglandins Leukot. Essent. Fatty Acids 2009, 80, 279–287. [Google Scholar] [CrossRef]
- Morimoto, M.; Reitz, R.C.; Morin, R.J.; Nguyen, K.; Ingelman-Sundberg, M.; French, S.W. Cyp-2E1 inhibitors partially ameliorate the changes in hepatic fatty acid composition induced in rats by chronic administration of ethanol and a high fat diet. J. Nutr. 1995, 125, 2953–2964. [Google Scholar]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Falck, J.R.; Harris, R.C. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J. Lipid Res. 2000, 41, 163–181. [Google Scholar]
- Rosell, M.; Hondares, E.; Iwamoto, S.; Gonzalez, F.J.; Wabitsch, M.; Staels, B.; Olmos, Y.; Monsalve, M.; Giralt, M.; Iglesias, R.; et al. Peroxisome proliferator-activated receptors-α and -γ, and camp-mediated pathways, control retinol-binding protein-4 gene expression in brown adipose tissue. Endocrinology 2012, 153, 1162–1173. [Google Scholar] [CrossRef]
- Melis, M.; Carta, G.; Pistis, M.; Banni, S. Physiological role of peroxisome proliferator-activated receptors type α on dopamine systems. CNS Neurol. Disord. Drug Targets 2013, 12, 70–77. [Google Scholar] [CrossRef]
- Fu, J.; Oveisi, F.; Gaetani, S.; Lin, E.; Piomelli, D. Oleoylethanolamide, an endogenous PPAR-α agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 2005, 48, 1147–1153. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Re, G.; Barbero, R.; Miolo, A.; Di Marzo, V. Palmitoylethanolamide, endocannabinoids and related cannabimimetic compounds in protection against tissue inflammation and pain: Potential use in companion animals. Vet. J. 2007, 173, 21–30. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carta, G.; Murru, E.; Cordeddu, L.; Ortiz, B.; Giordano, E.; Belury, M.A.; Quadro, L.; Banni, S. Metabolic Interactions between Vitamin A and Conjugated Linoleic Acid. Nutrients 2014, 6, 1262-1272. https://doi.org/10.3390/nu6031262
Carta G, Murru E, Cordeddu L, Ortiz B, Giordano E, Belury MA, Quadro L, Banni S. Metabolic Interactions between Vitamin A and Conjugated Linoleic Acid. Nutrients. 2014; 6(3):1262-1272. https://doi.org/10.3390/nu6031262
Chicago/Turabian StyleCarta, Gianfranca, Elisabetta Murru, Lina Cordeddu, Berenice Ortiz, Elena Giordano, Martha A. Belury, Loredana Quadro, and Sebastiano Banni. 2014. "Metabolic Interactions between Vitamin A and Conjugated Linoleic Acid" Nutrients 6, no. 3: 1262-1272. https://doi.org/10.3390/nu6031262
APA StyleCarta, G., Murru, E., Cordeddu, L., Ortiz, B., Giordano, E., Belury, M. A., Quadro, L., & Banni, S. (2014). Metabolic Interactions between Vitamin A and Conjugated Linoleic Acid. Nutrients, 6(3), 1262-1272. https://doi.org/10.3390/nu6031262