Vitamin A Derivatives as Treatment Options for Retinal Degenerative Diseases

Abstract
:
{kind=link}
{kind=link}
{kind=link}
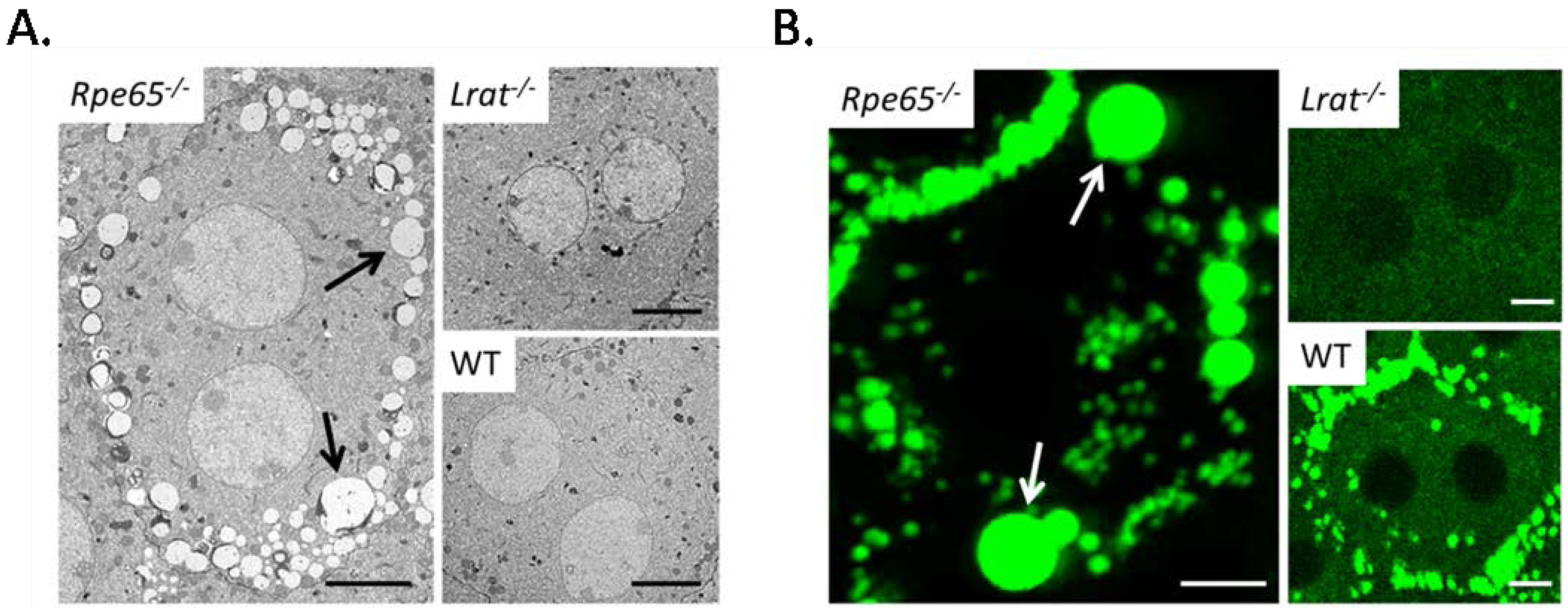{kind=link}
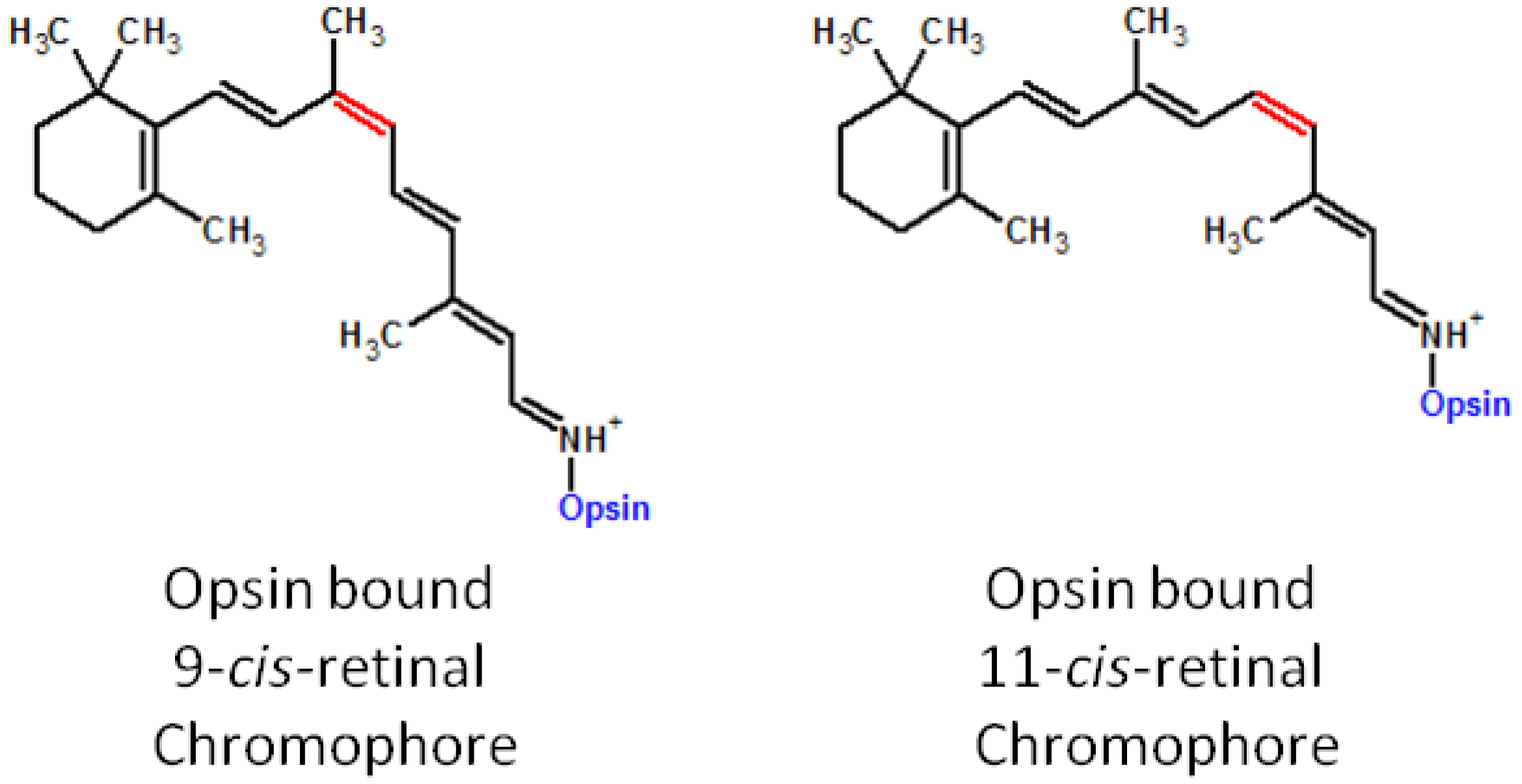{kind=link}
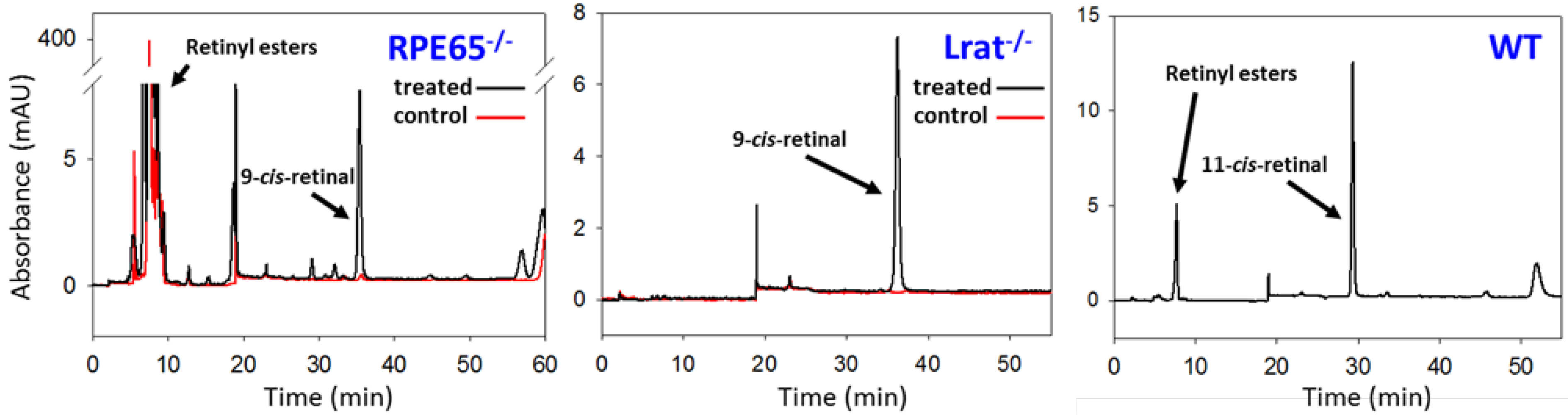{kind=link}
1. Introduction
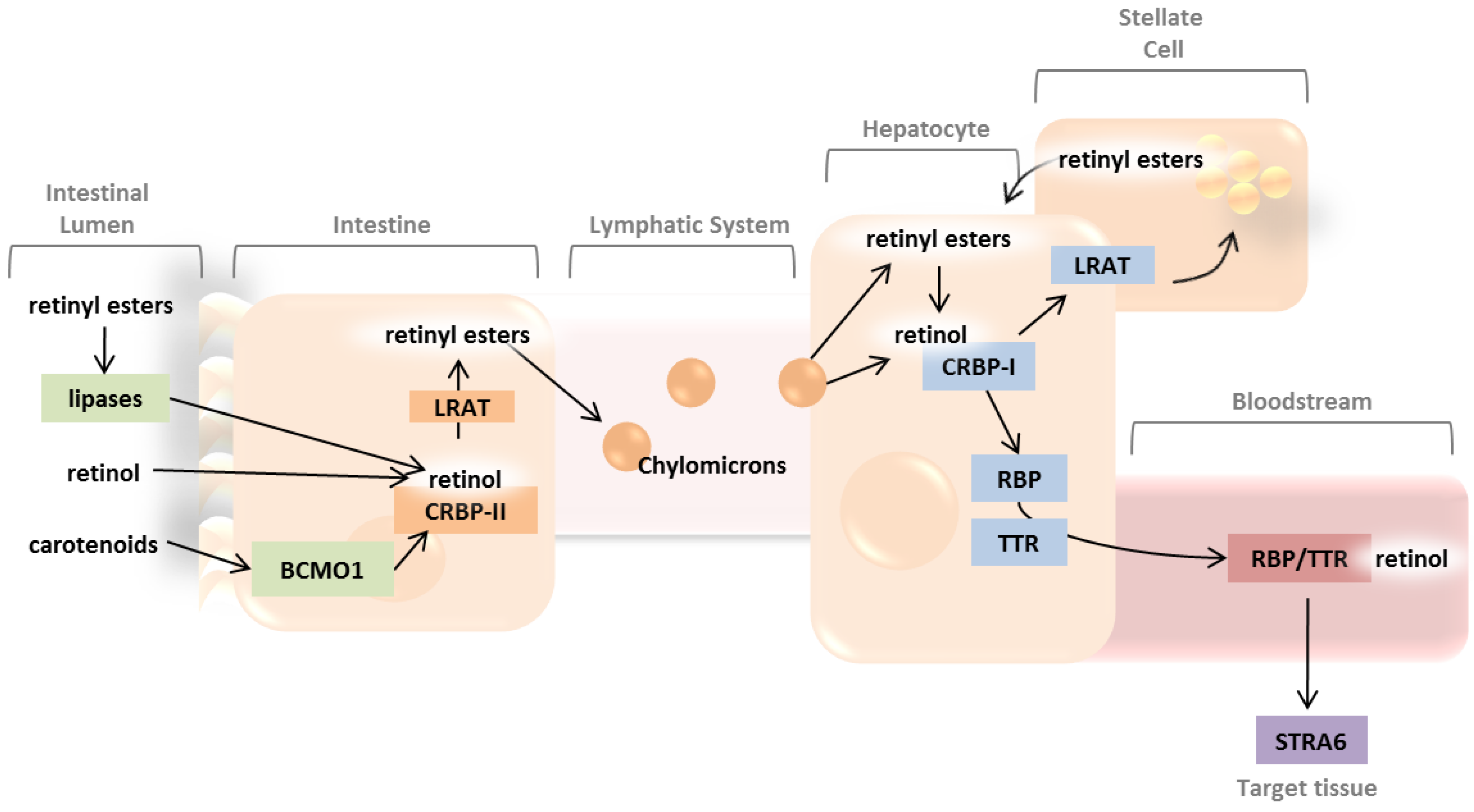
2. Absorption and Distribution of Dietary Vitamin A as Retinyl Esters and Provitamin A Carotenoids
2.1. Intestinal Uptake and Metabolism of Pro-Vitamin A Carotenoids
2.2. Intestinal Uptake of Retinyl Esters and Reesterification of Retinol by LRAT
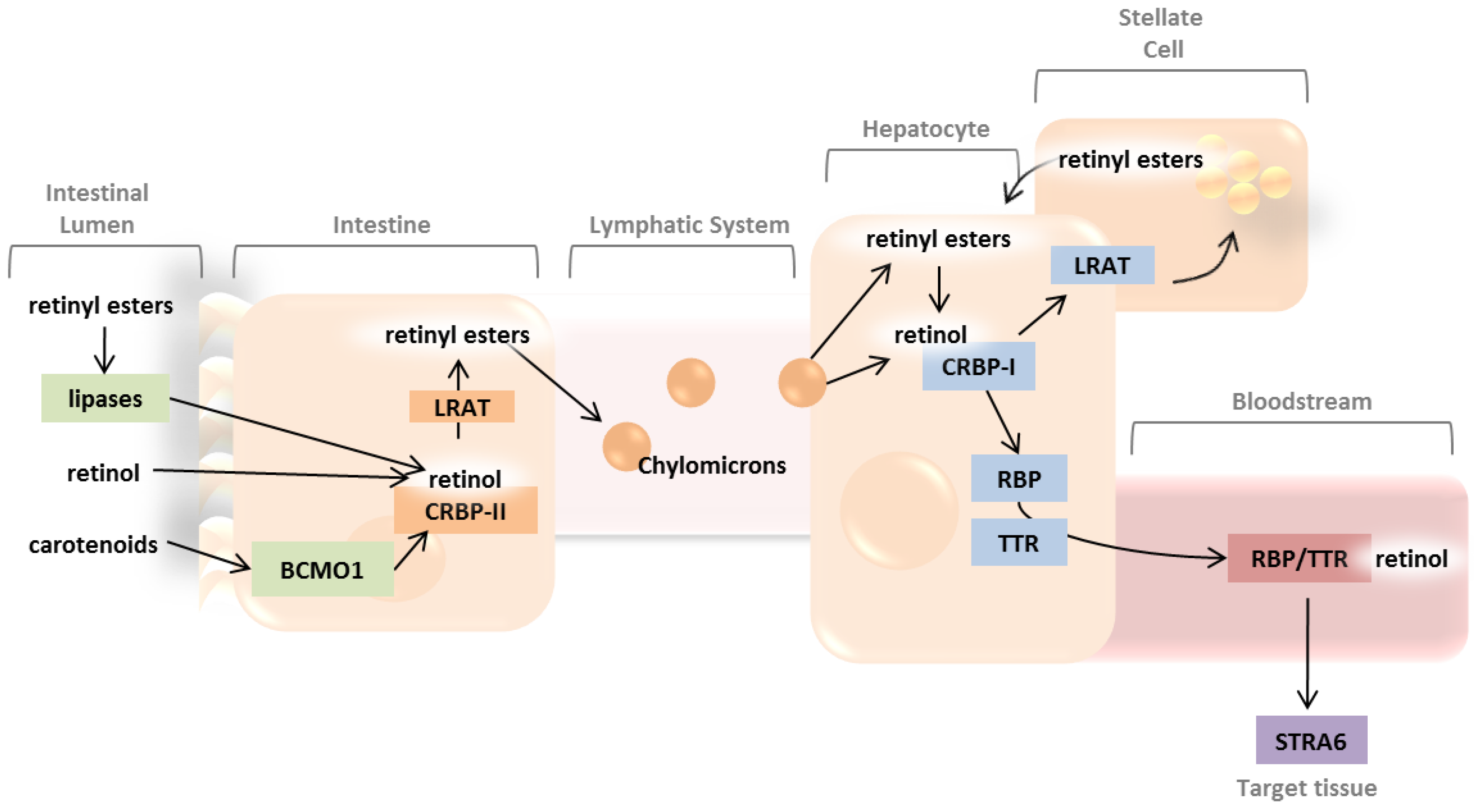
2.3. Systemic Circulation and Cellular Uptake by STRA6
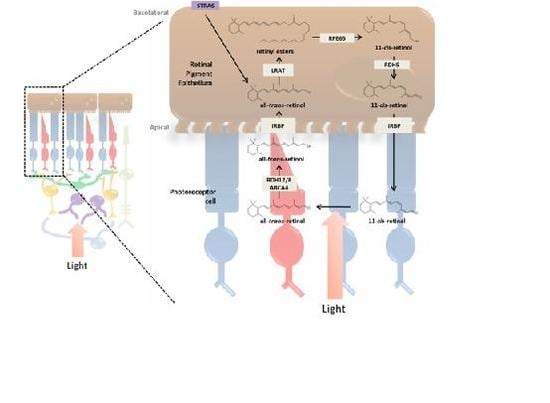
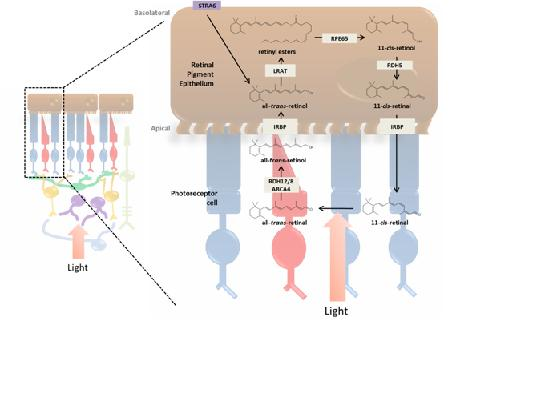
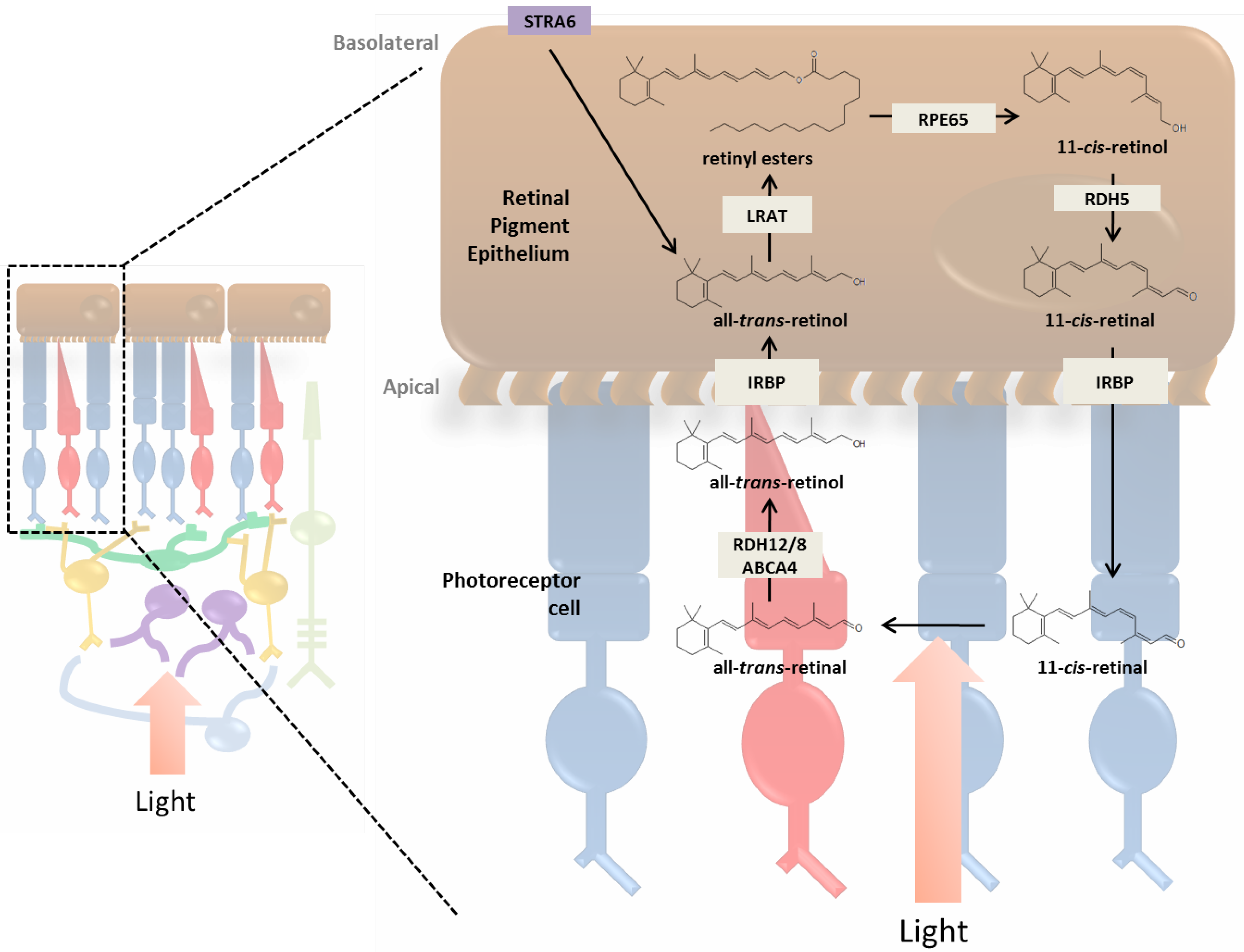
3. Incorporation of Retinol into the Retina and Visual Cycle
3.1. RPE and the Photoreceptor Visual Cycle
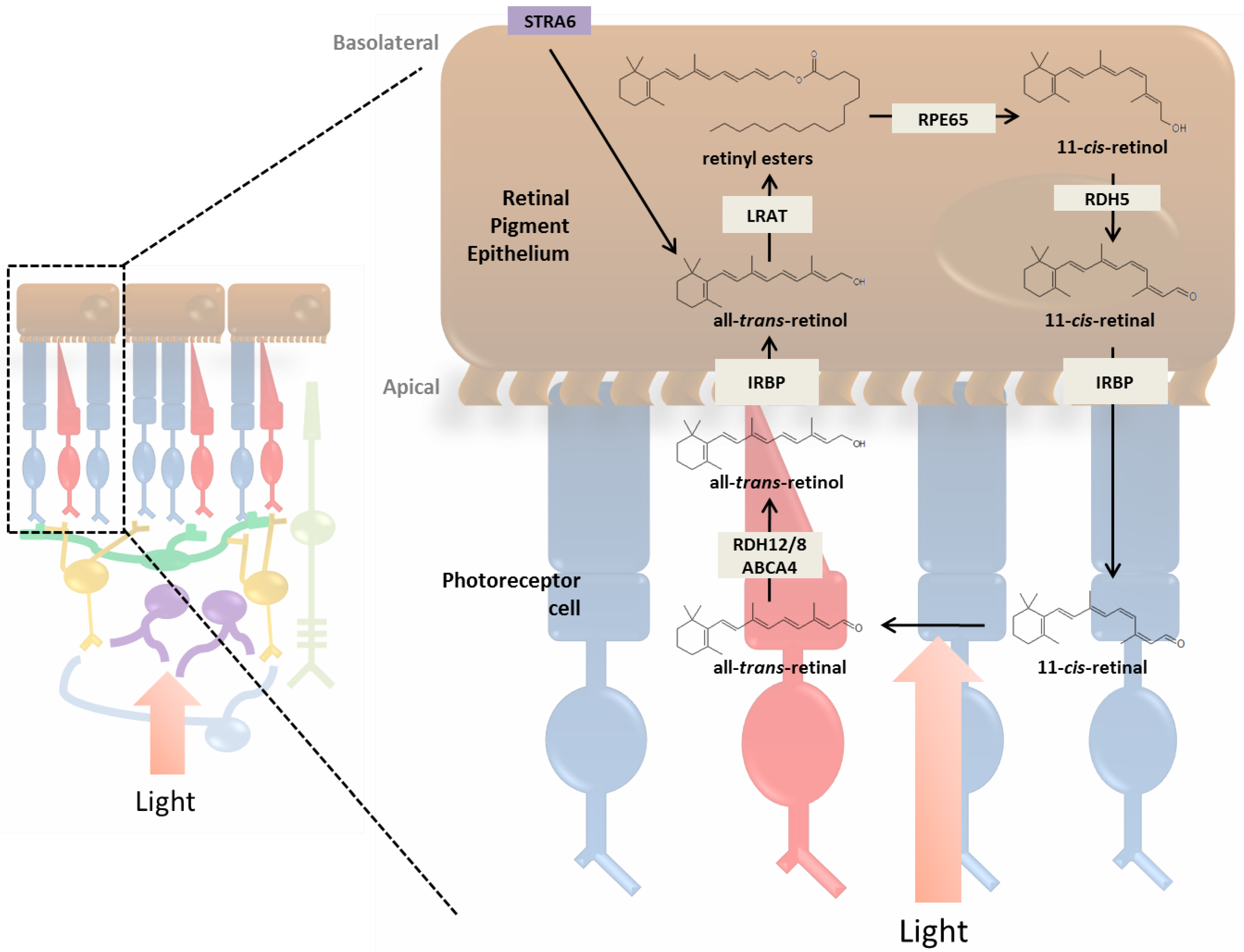
3.2. Müller Cells and the Cone Visual Cycle
3.3. Enzymatic Processing of Retinol in the RPE
3.4. Retinoid Transport between RPE and Photoreceptor Cells
3.5. Photoreceptor Cells and Visual Transduction
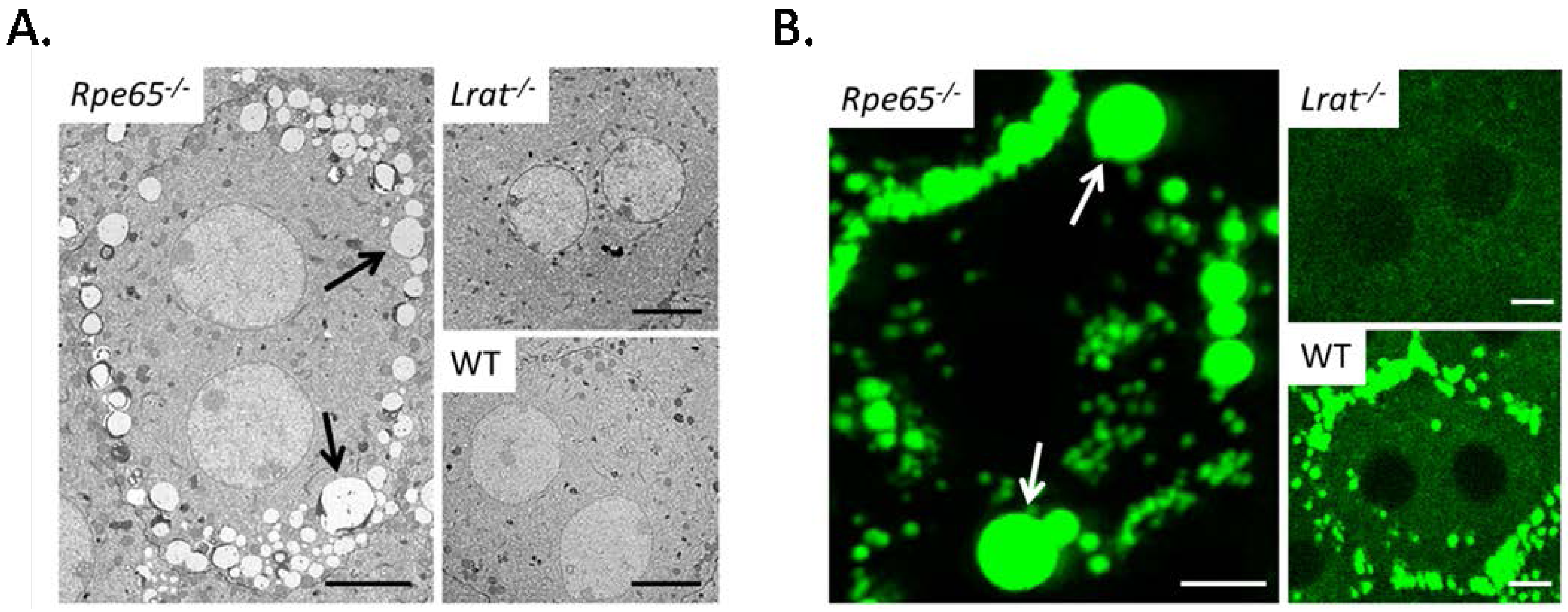
4. Deficiencies in 11-cis-Retinal and Associated Retinal Degenerative Diseases
4.1. Pathophysiology of 11-cis-Deficient Retinal Diseases in Mouse Models of Retinal Degeneration
5. Artificial Visual Chromophore Therapeutics and Further Applications
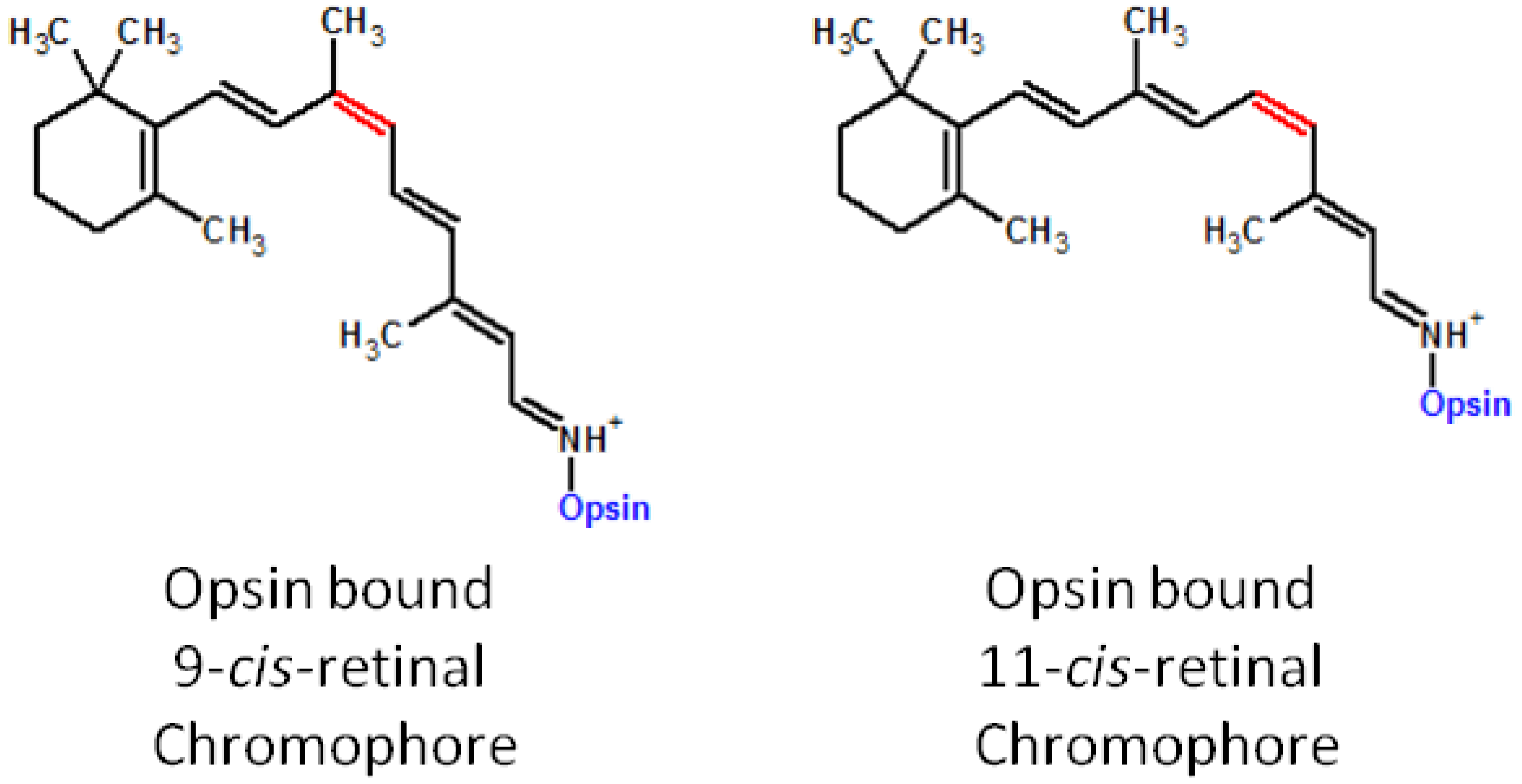
5.1. Prevention of the LCA Phenotype with Administration of 9-cis-Retinoids in Animal Models
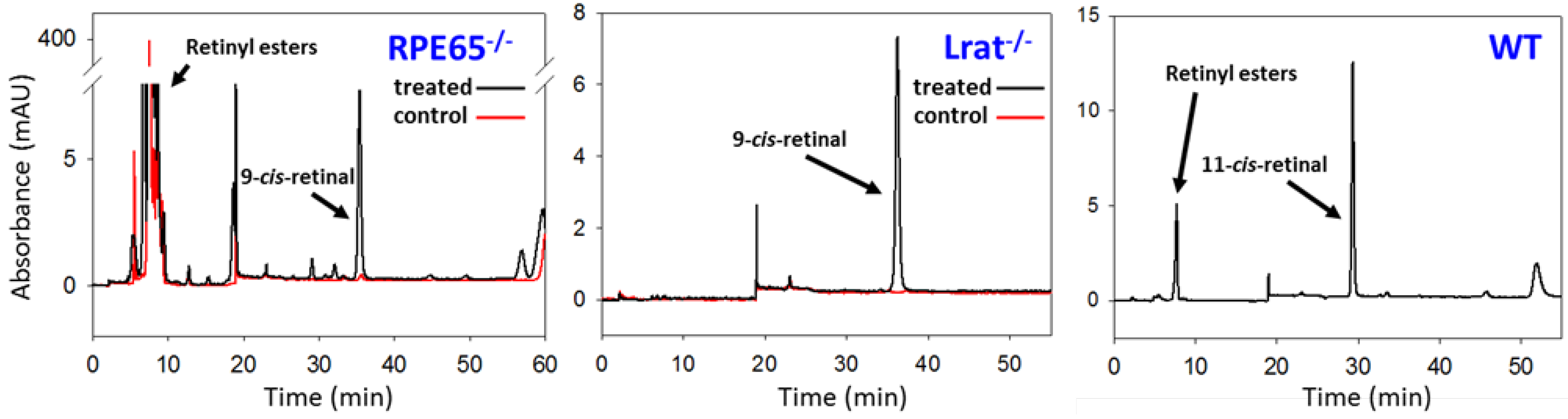
5.2. Therapeutics of 9-cis-Carotenoids in the Treatment of LCA
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Sommer, A.; Vyas, K.S. A global clinical view on vitamin A and carotenoids. Am. J. Clin. Nutr. 2012, 96, 1204S–1206S. [Google Scholar] [CrossRef]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Oren, T.; Sher, J.A.; Evans, T. Hematopoiesis and retinoids: Development and disease. Leuk. Lymphoma 2003, 44, 1881–1891. [Google Scholar] [CrossRef]
- Wald, G. Molecular basis of visual excitation. Science 1968, 162, 230–239. [Google Scholar]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef]
- Plack, P.A. Occurrence, absorption and distribution of vitamin A. Proc. Nutr. Soc. 1965, 24, 146–153. [Google Scholar] [CrossRef]
- Gaucheron, F. Milk and dairy products: A unique micronutrient combination. J. Am. Coll. Nutr. 2011, 30, 400S–409S. [Google Scholar] [CrossRef]
- Thurnham, D.I.; Northrop-Clewes, C.A. Optimal nutrition: Vitamin A and the carotenoids. Proc. Nutr. Soc. 1999, 58, 449–457. [Google Scholar] [CrossRef]
- Allen, L.H. To what extent can food-based approaches improve micronutrient status? Asia Pac. J. Clin. Nutr. 2008, 17, 103–105. [Google Scholar]
- Dew, S.E.; Ong, D.E. Specificity of the retinol transporter of the rat small intestine brush border. Biochemistry 1994, 33, 12340–12345. [Google Scholar] [CrossRef]
- Vachali, P.; Bhosale, P.; Bernstein, P.S. Microbial carotenoids. Methods Mol. Biol. 2012, 898, 41–59. [Google Scholar] [CrossRef]
- Bernstein, P.S.; Yoshida, M.D.; Katz, N.B.; McClane, R.W.; Gellermann, W. Raman detection of macular carotenoid pigments in intact human retina. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2003–2011. [Google Scholar]
- Moore, T. Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 1930, 24, 692–702. [Google Scholar]
- Amengual, J.; Lobo, G.P.; Golczak, M.; Li, H.N.; Klimova, T.; Hoppel, C.L.; Wyss, A.; Palczewski, K.; von Lintig, J. A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J. 2011, 25, 948–959. [Google Scholar] [CrossRef]
- Hu, K.Q.; Liu, C.; Ernst, H.; Krinsky, N.I.; Russell, R.M.; Wang, X.D. The biochemical characterization of ferret carotene-9′,10′-monooxygenase catalyzing cleavage of carotenoids in vitro and in vivo. J. Biol. Chem. 2006, 281, 19327–19338. [Google Scholar] [CrossRef]
- Lietz, G.; Oxley, A.; Boesch-Saadatmandi, C.; Kobayashi, D. Importance of beta,beta-carotene 15,15′-monooxygenase 1 (BCMO1) and beta,beta-carotene 9′,10′-dioxygenase 2 (BCDO2) in nutrition and health. Mol. Nutr. Food Res. 2012, 56, 241–250. [Google Scholar] [CrossRef]
- Lobo, G.P.; Hessel, S.; Eichinger, A.; Noy, N.; Moise, A.R.; Wyss, A.; Palczewski, K.; von Lintig, J. ISX is a retinoic acid-sensitive gatekeeper that controls intestinal β,β-carotene absorption and vitamin A production. FASEB J. 2010, 24, 1656–1666. [Google Scholar] [CrossRef]
- Seino, Y.; Miki, T.; Kiyonari, H.; Abe, T.; Fujimoto, W.; Kimura, K.; Takeuchi, A.; Takahashi, Y.; Oiso, Y.; Iwanaga, T.; et al. Isx participates in the maintenance of vitamin A metabolism by regulation of beta-carotene 15,15′-monooxygenase (Bcmo1) expression. J. Biol. Chem. 2008, 283, 4905–4911. [Google Scholar]
- O’Neill, M.E.; Thurnham, D.I. Intestinal absorption of beta-carotene, lycopene and lutein in men and women following a standard meal: Response curves in the triacylglycerol-rich lipoprotein fraction. Br. J. Nutr. 1998, 79, 149–159. [Google Scholar] [CrossRef]
- Rigtrup, K.M.; Kakkad, B.; Ong, D.E. Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: Probable identity with brush border phospholipase B. Biochemistry 1994, 33, 2661–2666. [Google Scholar] [CrossRef]
- Ong, D.E. Cellular transport and metabolism of vitamin A: Roles of the cellular retinoid-binding proteins. Nutr. Rev. 1994, 52, S24–S31. [Google Scholar] [CrossRef]
- Li, E.; Norris, A.W. Structure/function of cytoplasmic vitamin A-binding proteins. Annu. Rev. Nutr. 1996, 16, 205–234. [Google Scholar] [CrossRef]
- Newcomer, M.E.; Jamison, R.S.; Ong, D.E. Structure and function of retinoid-binding proteins. Subcell. Biochem. 1998, 30, 53–80. [Google Scholar] [CrossRef]
- Piantedosi, R.; Ghyselinck, N.; Blaner, W.S.; Vogel, S. Cellular retinol-binding protein type III is needed for retinoid incorporation into milk. J. Biol. Chem. 2005, 280, 24286–24292. [Google Scholar] [CrossRef]
- Blomhoff, R.; Green, M.H.; Berg, T.; Norum, K.R. Transport and storage of vitamin A. Science 1990, 250, 399–404. [Google Scholar] [CrossRef]
- Redgrave, T.G. Chylomicron metabolism. Biochem. Soc. Trans. 2004, 32, 79–82. [Google Scholar] [CrossRef]
- Goodman, D.W.; Huang, H.S.; Shiratori, T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. J. Lipid Res. 1965, 6, 390–396. [Google Scholar]
- Van Bennekum, A.M.; Kako, Y.; Weinstock, P.H.; Harrison, E.H.; Deckelbaum, R.J.; Goldberg, I.J.; Blaner, W.S. Lipoprotein lipase expression level influences tissue clearance of chylomicron retinyl ester. J. Lipid Res. 1999, 40, 565–574. [Google Scholar]
- MacDonald, P.N.; Ong, D.E. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J. Biol. Chem. 1988, 263, 12478–12482. [Google Scholar]
- O’Byrne, S.M.; Wongsiriroj, N.; Libien, J.; Vogel, S.; Goldberg, I.J.; Baehr, W.; Palczewski, K.; Blaner, W.S. Retinoid absorption and storage is impaired in mice lacking lecithin: Retinol acyltransferase (LRAT). J. Biol. Chem. 2005, 280, 35647–35657. [Google Scholar] [CrossRef]
- Orban, T.; Palczewska, G.; Palczewski, K. Retinyl ester storage particles (Retinosomes) from the retinal pigmented epithelium resemble lipid droplets in other tissues. J. Biol. Chem. 2011, 286, 17248–17258. [Google Scholar]
- Frey, S.K.; Vogel, S. Vitamin A metabolism and adipose tissue biology. Nutrients 2011, 3, 27–39. [Google Scholar] [CrossRef]
- Chytil, F. The lungs and vitamin A. Am. J. Physiol. 1992, 262, L517–L527. [Google Scholar]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef]
- Seeliger, M.W.; Biesalski, H.K.; Wissinger, B.; Gollnick, H.; Gielen, S.; Frank, J.; Beck, S.; Zrenner, E. Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3–11. [Google Scholar]
- Wolf, G. Retinol transport and metabolism in transthyretin-“knockout” mice. Nutr. Rev. 1995, 53, 98–99. [Google Scholar] [CrossRef]
- Zanotti, G.; Berni, R. Plasma retinol-binding protein: Structure and interactions with retinol, retinoids, and transthyretin. Vitam. Horm. 2004, 69, 271–295. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef]
- White, T.; Lu, T.Y.; Metlapally, R.; Katowitz, J.; Kherani, F.; Wang, T.Y.; Tran-Viet, K.N.; Young, T.L. Identification of STRA6 and SKI sequence variants in patients with anophthalmia/microphthalmia. Mol. Vis. 2008, 14, 2458–2465. [Google Scholar]
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039. [Google Scholar] [CrossRef]
- Saari, J.C. Vitamin A metabolism in rod and cone visual cycles. Annu. Rev. Nutr. 2012, 32, 125–145. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Golczak, M.; Moise, A.R.; Palczewski, K. Aberrant metabolites in mouse models of congenital blinding diseases, formation and storage of retinyl esters. Biochemistry 2006, 45, 4210–4219. [Google Scholar] [CrossRef]
- Imanishi, Y.; Batten, M.L.; Piston, D.W.; Baehr, W.; Palczewski, K. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. J. Cell Biol. 2004, 164, 373–383. [Google Scholar] [CrossRef]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol.Chem. 2004, 279, 10422–10432. [Google Scholar]
- Gollapalli, D.R.; Rando, R.R. All-trans-retinyl esters are the substrates for isomerization in the vertebrate visual cycle. Biochemistry 2003, 42, 5809–5818. [Google Scholar] [CrossRef]
- Wang, J.S.; Estevez, M.E.; Cornwall, M.C.; Kefalov, V.J. Intra-retinal visual cycle required for rapid and complete cone dark adaptation. Nat. Neurosci. 2009, 12, 295–302. [Google Scholar] [CrossRef]
- Jones, G.J.; Crouch, R.K.; Wiggert, B.; Cornwall, M.C.; Chader, G.J. Retinoid requirements for recovery of sensitivity after visual-pigment bleaching in isolated photoreceptors. Proc. Natl. Acad. Sci. USA 1989, 86, 9606–9610. [Google Scholar]
- Kefalov, V.J. Rod and cone visual pigments and phototransduction through pharmacological, genetic, and physiological approaches. J. Biol. Chem. 2012, 287, 1635–1641. [Google Scholar] [CrossRef]
- Wang, J.S.; Kefalov, V.J. The cone-specific visual cycle. Prog. Retin. Eye Res. 2011, 30, 115–128. [Google Scholar] [CrossRef]
- Trehan, A.; Canada, F.J.; Rando, R.R. Inhibitors of retinyl ester formation also prevent the biosynthesis of 11-cis-retinol. Biochemistry 1990, 29, 309–312. [Google Scholar] [CrossRef]
- Redmond, T.M.; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.X.; Crouch, R.K.; Pfeifer, K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar] [CrossRef]
- Parker, R.O.; Crouch, R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010, 91, 788–792. [Google Scholar] [CrossRef]
- Rohrer, B.; Lohr, H.R.; Humphries, P.; Redmond, T.M.; Seeliger, M.W.; Crouch, R.K. Cone opsin mislocalization in Rpe65(−/−) mice: A defect that can be corrected by 11-cis retinal. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3876–3882. [Google Scholar] [CrossRef]
- Driessen, C.A.; Winkens, H.J.; Hoffmann, K.; Kuhlmann, L.D.; Janssen, B.P.; van Vugt, A.H.; van Hooser, J.P.; Wieringa, B.E.; Deutman, A.F.; Palczewski, K.; et al. Disruption of the 11-cis-retinol dehydrogenase gene leads to accumulation of cis-retinols and cis-retinyl esters. Mol. Cell. Biol. 2000, 20, 4275–4287. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, F. Interphotoreceptor retinoid-binding protein—An old gene for new eyes. Vis. Res. 2003, 43, 3021–3036. [Google Scholar] [CrossRef]
- Wu, Q.; Blakeley, L.R.; Cornwall, M.C.; Crouch, R.K.; Wiggert, B.N.; Koutalos, Y. Interphotoreceptor retinoid-binding protein is the physiologically relevant carrier that removes retinol from rod photoreceptor outer segments. Biochemistry 2007, 46, 8669–8679. [Google Scholar] [CrossRef]
- Jin, M.; Li, S.; Nusinowitz, S.; Lloyd, M.; Hu, J.; Radu, R.A.; Bok, D.; Travis, G.H. The role of interphotoreceptor retinoid-binding protein on the translocation of visual retinoids and function of cone photoreceptors. J. Neurosci. 2009, 29, 1486–1495. [Google Scholar]
- Parker, R.O.; Fan, J.; Nickerson, J.M.; Liou, G.I.; Crouch, R.K. Normal cone function requires the interphotoreceptor retinoid binding protein. J. Neurosci. 2009, 29, 4616–4621. [Google Scholar] [CrossRef]
- Wisard, J.; Faulkner, A.; Chrenek, M.A.; Waxweiler, T.; Waxweiler, W.; Donmoyer, C.; Liou, G.I.; Craft, C.M.; Schmid, G.F.; Boatright, J.H.; et al. Exaggerated eye growth in IRBP-deficient mice in early development. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5804–5811. [Google Scholar] [CrossRef]
- Garlipp, M.A.; Gonzalez-Fernandez, F. Cone outer segment and Muller microvilli pericellular matrices provide binding domains for interphotoreceptor retinoid-binding protein (IRBP). Exp. Eye Res. 2013. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, F. Evolution of the visual cycle: The role of retinoid-binding proteins. J. Endocrinol. 2002, 175, 75–88. [Google Scholar] [CrossRef]
- Matsumoto, H.; Yoshizawa, T. Existence of a beta-ionone ring-binding site in the rhodopsin molecule. Nature 1975, 258, 523–526. [Google Scholar] [CrossRef]
- Bownds, D. Site of attachment of retinal in rhodopsin. Nature 1967, 216, 1178–1181. [Google Scholar] [CrossRef]
- Jastrzebska, B.; Debinski, A.; Filipek, S.; Palczewski, K. Role of membrane integrity on G protein-coupled receptors: Rhodopsin stability and function. Prog. Lipid Res. 2011, 50, 267–277. [Google Scholar] [CrossRef]
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef]
- Wassle, H. Parallel processing in the mammalian retina. Nat. Rev. Neurosci. 2004, 5, 747–757. [Google Scholar] [CrossRef]
- Carleton, K.L.; Spady, T.C.; Cote, R.H. Rod and cone opsin families differ in spectral tuning domains but not signal transducing domains as judged by saturated evolutionary trace analysis. J. Mol. Evol. 2005, 61, 75–89. [Google Scholar] [CrossRef]
- Saari, J.C.; Nawrot, M.; Garwin, G.G.; Kennedy, M.J.; Hurley, J.B.; Ghyselinck, N.B.; Chambon, P. Analysis of the visual cycle in cellular retinol-binding protein type I (CRBPI) knockout mice. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1730–1735. [Google Scholar]
- Rozanowska, M.; Handzel, K.; Boulton, M.E.; Różanowski, B. Cytotoxicity of all-trans-retinal increases upon photodegradation. Photochem. Photobiol. 2012, 88, 1362–1372. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Chou, S.; Desai, A.; Hoppel, C.L.; Matsuyama, S.; Palczewski, K. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem. 2009, 284, 15173–15183. [Google Scholar]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal-pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Znoiko, S.L.; Rohrer, B.; Lu, K.; Lohr, H.R.; Crouch, R.K.; Ma, J.X. Downregulation of cone-specific gene expression and degeneration of cone Photoreceptors in the Rpe65(−/−) mouse at early ages. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1473–1479. [Google Scholar] [CrossRef]
- Fan, J.; Rohrer, B.; Frederick, J.M.; Baehr, W.; Crouch, R.K. Rpe65(−/−) and Lrat(−/−) mice: Comparable models of Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2384–2389. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, J.; Li, S.; Karan, S.; Rohrer, B.; Palczewski, K.; Frederick, J.M.; Crouch, R.K.; Baehr, W. Trafficking of membrane-associated proteins to cone photoreceptor outer segments requires the chromophore 11-cis-retinal. J. Neurosci. 2008, 28, 4008–4014. [Google Scholar]
- Zhang, T.; Baehr, W.; Fu, Y. Chemical chaperone TUDCA preserves cone photoreceptors in a mouse model of Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3349–3356. [Google Scholar] [CrossRef]
- Sato, K.; Ozaki, T.; Ishiguro, S.; Nakazawa, M. M-opsin protein degradation is inhibited by MG-132 in Rpe65(−/−) retinal explant culture. Mol. Vis. 2012, 18, 1516–1525. [Google Scholar]
- Batten, M.L.; Imanishi, Y.; Yu, D.C.; Doan, T.; Zhu, L.; Pang, J.J.; Glushakova, L.; Moise, A.R.; Baehr, W.; van Gelder, R.N.; et al. Parmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005, 2, e333. [Google Scholar] [CrossRef]
- Deretic, D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vis. Res. 2006, 46, 4427–4433. [Google Scholar] [CrossRef]
- Noorwez, S.M.; Malhotra, R.; McDowell, J.H.; Smith, K.A.; Krebs, M.P.; Kaushal, S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J. Biol. Chem. 2004, 279, 16278–16284. [Google Scholar]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef]
- Liu, L.; Gudas, L.J. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J. Biol. Chem. 2005, 280, 40226–40234. [Google Scholar] [CrossRef]
- Perrault, I.; Rozet, J.R.; Gerber, S.; Ghazi, I.; Leowski, C.; Ducroq, D.; Souied, E.; Dufier, J.L.; Munnich, A.; Kaplan, J. Leber congenital amaurosis. Mol. Genet. Metab. 1999, 68, 200–208. [Google Scholar] [CrossRef]
- Hufnagel, R.B.; Ahmed, Z.M.; Corrêa, Z.M.; Sisk, R.A. Gene therapy for Leber congenital amaurosis: Advances and future directions. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 1117–1128. [Google Scholar] [CrossRef]
- Perrault, I.; Hanein, S.; Gerber, S.; Barbet, F.; Dufier, J.L.; Munnich, A.; Rozet, J.M.; Kaplan, J. Evidence of autosomal dominant Leber congenital amaurosis (LCA) underlain by a CRX heterozygous null allele. J. Med. Genet. 2003, 40, e90. [Google Scholar] [CrossRef]
- Den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar]
- Aleman, T.S.; Jacobson, S.G.; Chico, J.D.; Scott, M.L.; Cheung, A.Y.; Windsor, E.A.; Furushima, M.; Redmond, T.M.; Bennett, J.; Palczewski, K; et al. Impairment of the transient pupillary light reflex in Rpe65(−/−) mice and humans with leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1259–1271. [Google Scholar] [CrossRef]
- Lorenz, B.; Gyürüs, P.; Preising, M.; Bremser, D.; Gu, S.; Andrassi, M.; Gerth, C.; Gal, A. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Investig. Ophthalmol.Vis. Sci. 2000, 41, 2735–2742. [Google Scholar]
- Van Hooser, J.P.; Aleman, T.S.; He, Y.G.; Cideciyan, A.V.; Kuksa, V.; Pittler, S.J.; Stone, E.M.; Jacobson, S.G.; Palczewski, K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc. Natl. Acad. Sci. USA 2000, 97, 8623–8628. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Van Hooser, J.P.; Liang, Y.; Maeda, T.; Kuksa, V.; Jang, G.F.; He, Y.G.; Rieke, F.; Fong, H.K.; Detwiler, P.B.; Palczewski, K. Recovery of visual functions in a mouse model of Leber congenital amaurosis. J. Biol. Chem. 2002, 277, 19173–19182. [Google Scholar] [CrossRef]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le, T.I.; Teller, D.C.; Okada, T.; Stenkamp, R.E; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef]
- Palczewski, K. Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 2010, 31, 284–295. [Google Scholar] [CrossRef]
- Gao, S.Q.; Maeda, T.; Okano, K.; Palczewski, K. A microparticle/hydrogel combination drug-delivery system for sustained release of retinoids. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6314–6323. [Google Scholar] [CrossRef]
- Gearhart, P.M.; Gearhart, C.; Thompson, D.A.; Petersen-Jones, S.M. Improvement of visual performance with intravitreal administration of 9-cis-retinal in Rpe65-mutant dogs. Arch. Ophthalmol. 2010, 128, 1442–1448. [Google Scholar] [CrossRef]
- Collins, M.D.; Tzimas, G.; Hummler, H.; Bürgin, H.; Nau, H. Comparative teratology and transplacental pharmacokinetics of all-trans-retinoic acid, 13-cis-retinoic acid, and retinyl palmitate following daily administrations in rats. Toxicol. Appl. Pharmacol. 1994, 127, 132–144. [Google Scholar] [CrossRef]
- Maeda, T.; Maeda, A.; Casadesus, G.; Palczewski, K.; Margaron, P. Evaluation of 9-cis-retinyl acetate therapy in Rpe65−/− mice. Investig. Ophthalmol.Vis. Sci. 2009, 50, 4368–4378. [Google Scholar] [CrossRef]
- Qtaishat, N.M.; Redmond, T.M.; Pepperberg, D.R. Acute radiolabeling of retinoids in eye tissues of normal and rpe65-deficient mice. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1435–1446. [Google Scholar] [CrossRef]
- Maeda, T.; Dong, Z.; Jin, H.; Sawada, O.; Gao, S.; Utkhede, D.; Monk, W.; Palczewska, G.; Palczewski, K. QLT091001, a 9-cis-retinal analog, is well-tolerated by retinas of mice with impaired visual cycles. Investig. Ophthalmol. Vis. Sci. 2013, 54, 455–466. [Google Scholar] [CrossRef]
- Maeda, T.; Cideciyan, A.V.; Maeda, A.; Golczak, M.; Aleman, T.S.; Jacobson, S.G.; Palczewski, K. Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro-drug, 9-cis-retinyl acetate. Hum. Mol. Genet. 2009, 18, 2277–2287. [Google Scholar] [CrossRef]
- Hamann, S.; Schorderet, D.F.; Cottet, S. Bax-induced apoptosis in leber’s congenital amaurosis: A dual role in rod and cone degeneration. PLoS One 2009, 4, e6616. [Google Scholar] [CrossRef]
- Baxi, S.C.; Dailey, G.E. Hypervitaminosis A—A Cause of Hypercalcemia. West. J. Med. 1982, 137, 429–431. [Google Scholar]
- Guarascio, P.; Portmann, B.; Visco, G.; Williams, R. Liver damage with reversible portal-hypertension from vitamin A intoxication: Demonstration of ito cells. J. Clin. Pathol. 1983, 36, 769–771. [Google Scholar] [CrossRef]
- Nollevaux, M.C.; Guiot, Y.; Horsmans, Y.; Leclercq, I.; Rahier, J.; Geubel, A.P.; Sempoux, C. Hypervitaminosis A-induced liver fibrosis: Stellate cell activation and daily dose consumption. Liver Int. 2006, 26, 182–186. [Google Scholar] [CrossRef]
- Jirillo, E.; Jirillo, F.; Magrone, T. Healthy effects exerted by prebiotics, probiotics, and symbiotics with special reference to their impact on the immune system. Int. J. Vitam. Nutr. Res. 2012, 82, 200–208. [Google Scholar] [CrossRef]
- Soprano, D.R.; Soprano, K.J. Retinoids as teratogens. Annu. Rev. Nutr. 1995, 15, 111–132. [Google Scholar] [CrossRef]
- Collins, M.D.; Mao, G.E. Teratology of retinoids. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 399–430. [Google Scholar] [CrossRef]
- Rothman, K.J.; Moore, L.L.; Singer, M.R.; Nguyen, U.D.T.; Mannino, S.; Milunsky, A. Teratogenicity of high vitamin A intake. N. Engl. J. Med. 1995, 333, 1369–1373. [Google Scholar] [CrossRef]
- Lobo, G.P.; Amengual, J.; Baus, D.; Shivdasani, R.A.; Taylor, D.; von Lintig, J. Genetics and diet regulate vitamin A production via the homeobox transcription factor ISX. J. Biol. Chem. 2013, 288, 9017–9027. [Google Scholar]
- Rotenstreich, Y.; Harats, D.; Shaish, A.; Pras, E.; Belkin, M. Treatment of a retinal dystrophy, fundus albipunctatus, with oral 9-cis-β-carotene. Br. J. Ophthalmol. 2010, 94, 616–621. [Google Scholar] [CrossRef]
- Ajmal, M.; Khan, M.I.; Neveling, K.; Khan, Y.M.; Ali, S.H.; Ahmed, W.; Iqbal, M.S.; Azam, M.; den Hollander, A.I.; Collin, R.W.; et al. Novel mutations in RDH5 cause fundus albipunctatus in two consanguineous Pakistani families. Mol. Vis. 2012, 18, 1558–1571. [Google Scholar]
- Maeda, T.; Perusek, L.; Amengual, J.; Babino, D.; Palczewski, K.; von Lintig, J. Dietary 9-cis-β,β-carotene fails to rescue vision in mouse models of leber congenital amaurosis. Mol. Pharmacol. 2011, 80, 943–952. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perusek, L.; Maeda, T. Vitamin A Derivatives as Treatment Options for Retinal Degenerative Diseases. Nutrients 2013, 5, 2646-2666. https://doi.org/10.3390/nu5072646
Perusek L, Maeda T. Vitamin A Derivatives as Treatment Options for Retinal Degenerative Diseases. Nutrients. 2013; 5(7):2646-2666. https://doi.org/10.3390/nu5072646
Chicago/Turabian StylePerusek, Lindsay, and Tadao Maeda. 2013. "Vitamin A Derivatives as Treatment Options for Retinal Degenerative Diseases" Nutrients 5, no. 7: 2646-2666. https://doi.org/10.3390/nu5072646
APA StylePerusek, L., & Maeda, T. (2013). Vitamin A Derivatives as Treatment Options for Retinal Degenerative Diseases. Nutrients, 5(7), 2646-2666. https://doi.org/10.3390/nu5072646