Fat Depots, Free Fatty Acids, and Dyslipidemia

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Adipose Tissue and Free Fatty Acids
3. Fat Depots
4. Obesity, Fat Depots, and FFA
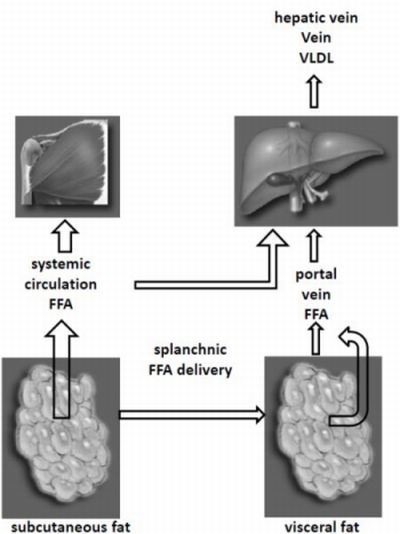
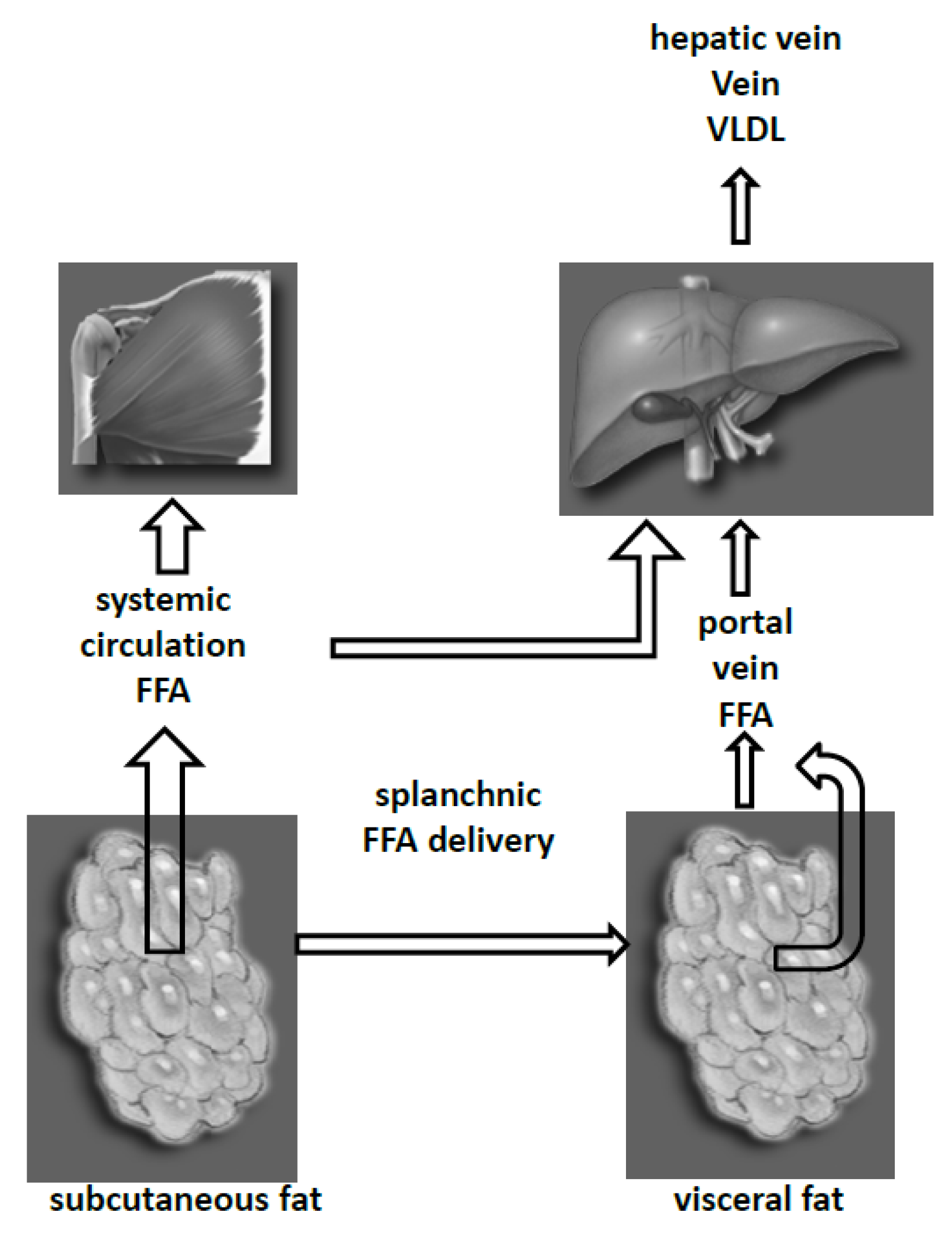
5. Fat Depots and Liver FFA Metabolism
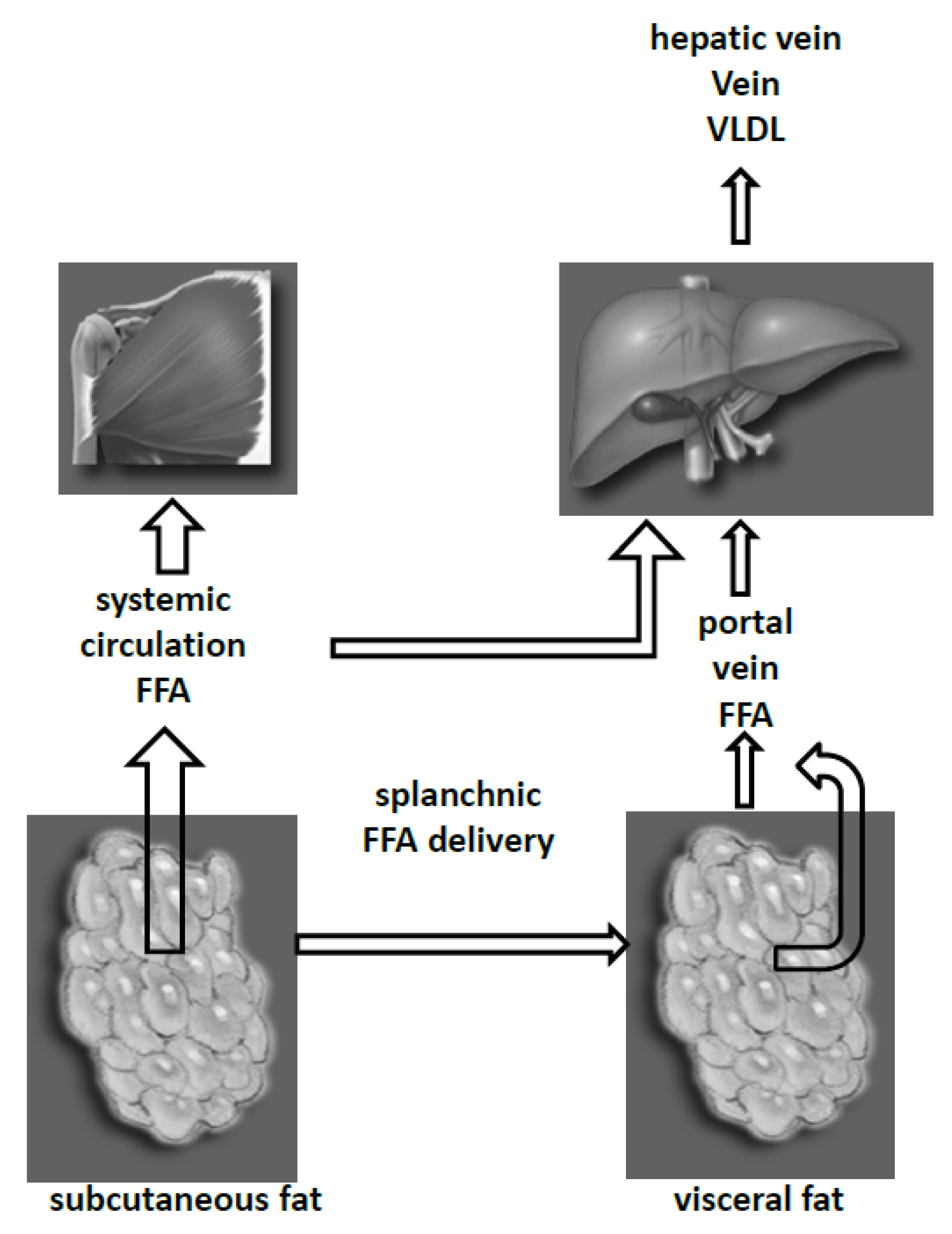
6. Fat Depots VLDL and LDL
7. Fat Depots and HDL Cholesterol
8. Conclusions
Conflict of Interest
References
- Lakka, T.A.; Lakka, H.M.; Salonen, R.; Kaplan, G.A.; Salonen, J.T. Abdominal obesity is associated with accelerated progression of carotid atherosclerosis in men. Atherosclerosis 2001, 154, 497–504. [Google Scholar] [CrossRef]
- Lakka, H.M.; Lakka, T.A.; Tuomilehto, J.; Salonen, J.T. Abdominal obesity is associated with increased risk of acute coronary events in men. Eur. Heart J. 2002, 23, 706–713. [Google Scholar] [CrossRef]
- Despres, J.P.; Moorjani, S.; Lupien, P.J.; Tremblay, A.; Nadeau, A.; Bouchard, C. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 1990, 10, 497–511. [Google Scholar] [CrossRef]
- Jensen, M.D. Role of body fat distribution and the metabolic complications of obesity. J. Clin. Endocrinol. Metab. 2008, 93, S57–S63. [Google Scholar] [CrossRef]
- Bjorntorp, P. “Portal” adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis 1990, 10, 493–496. [Google Scholar] [CrossRef]
- Kissebah, A.H.; Peiris, A.N. Biology of regional body fat distribution: Relationship to non-insulin-dependent diabetes mellitus. Diabetes Metab. Rev. 1989, 5, 83–109. [Google Scholar]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Gertz, E.W.; Neese, R.A.; Mayr, M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J. Clinical Investig. 1987, 79, 359–366. [Google Scholar] [CrossRef]
- Havel, R.J.; Kane, J.P.; Balasse, E.O.; Segel, N.; Basso, L.V. Splanchnic metabolism of free fatty acids and production of triglycerides of very low density lipoproteins in normotriglyceridemic and hypertriglyceridemic humans. J. Clin. Invest. 1970, 49, 2017–2035. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141. [Google Scholar]
- Byrne, C.D.; Wareham, N.J.; Brown, D.C.; Clark, P.M.; Cox, L.J.; Day, N.E.; Palmer, C.R.; Wang, T.W.; Williams, D.R.; Hales, C.N. Hypertriglyceridaemia in subjects with normal and abnormal glucose tolerance: Relative contributions of insulin secretion, insulin resistance and suppression of plasma non-esterified fatty acids. Diabetologia 1994, 37, 889–896. [Google Scholar] [CrossRef]
- Lewis, G.F.; O’Meara, N.M.; Soltys, P.A.; Blackman, J.D.; Iverius, P.H.; Pugh, W.L.; Getz, G.S.; Polonsky, K.S. Fasting hypertriglyceridemia in noninsulin-dependent diabetes mellitus is an important predictor of postprandial lipid and lipoprotein abnormalities. J. Clin. Endocrinol. Metab. 1991, 72, 934–944. [Google Scholar] [CrossRef]
- McKeigue, P.M.; Laws, A.; Chen, Y.D.; Marmot, M.G.; Reaven, G.M. Relation of plasma triglyceride and apoB levels to insulin-mediated suppression of nonesterified fatty acids. Possible explanation for sex differences in lipoprotein pattern. Arterioscler. Thromb. 1993, 13, 1187–1192. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H.; Taskinen, M.R. Interrelationships among insulin’s antilipolytic and glucoregulatory effects and plasma triglycerides in nondiabetic and diabetic patients with endogenous hypertriglyceridemia. Diabetes 1988, 37, 1271–1278. [Google Scholar]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006, 113, 898–918. [Google Scholar] [CrossRef]
- Goossens, G.H. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol. Behav. 2008, 94, 206–218. [Google Scholar] [CrossRef]
- Frayn, K.N. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar]
- Tchoukalova, Y.D.; Koutsari, C.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Thomou, T.; Kirkland, J.L.; Jensen, M.D. Sex- and depot-dependent differences in adipogenesis in normal-weight humans. Obesity (Silver Spring) 2010, 18, 1875–1880. [Google Scholar] [CrossRef]
- Kelley, D.E.; Thaete, F.L.; Troost, F.; Huwe, T.; Goodpaster, B.H. Subdivisions of subcutaneous abdominal adipose tissue and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E941–E948. [Google Scholar]
- Jensen, M.D.; Sarr, M.G.; Dumesic, D.A.; Southorn, P.A.; Levine, J.A. Regional uptake of meal fatty acids in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1282–E1288. [Google Scholar]
- Rosito, G.A.; Massaro, J.M.; Hoffmann, U.; Ruberg, F.L.; Mahabadi, A.A.; Vasan, R.S.; O'Donnell, C.J.; Fox, C.S. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community-based sample: The Framingham Heart Study. Circulation 2008, 117, 605–613. [Google Scholar] [CrossRef]
- Birkenhager, J.C.; Tjabbes, T. Turnover rate of plasma FFA and rate of esterification of plasma FFA to plasma triglycerides in obese humans before and after weight reduction. Metabolism 1969, 18, 18–32. [Google Scholar] [CrossRef]
- Campbell, P.J.; Carlson, M.G.; Nurjhan, N. Fat metabolism in human obesity. Am. J. Physiol. 1994, 266, E600–E605. [Google Scholar]
- Horowitz, J.F.; Coppack, S.W.; Paramore, D.; Cryer, P.E.; Zhao, G.; Klein, S. Effect of short-term fasting on lipid kinetics in lean and obese women. Am. J. Physiol. 1999, 276, E278–E284. [Google Scholar]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; Karpe, F. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar] [CrossRef]
- Kissebah, A.H.; Krakower, G.R. Regional adiposity and morbidity. Physiol. Rev. 1994, 74, 761–811. [Google Scholar]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal visceral and subcutaneous adipose tissue compartments: Association with metabolic risk factors in the Framingham Heart Study. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef]
- Manolopoulos, K.N.; Karpe, F.; Frayn, K.N. Gluteofemoral body fat as a determinant of metabolic health. Int. J. Obes. (Lond.) 2010, 34, 949–959. [Google Scholar] [CrossRef]
- Snijder, M.B.; Zimmet, P.Z.; Visser, M.; Dekker, J.M.; Seidell, J.C.; Shaw, J.E. Independent and opposite associations of waist and hip circumferences with diabetes, hypertension and dyslipidemia: The AusDiab Study. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 402–409. [Google Scholar]
- Nielsen, S.; Guo, Z.; Albu, J.B.; Klein, S.; O’Brien, P.C.; Jensen, M.D. Energy expenditure, sex, and endogenous fuel availability in humans. J. Clin. Invest. 2003, 111, 981–988. [Google Scholar]
- Saloranta, C.; Franssila-Kallunki, A.; Ekstrand, A.; Taskinen, M.R.; Groop, L. Modulation of hepatic glucose production by non-esterified fatty acids in type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1991, 34, 409–415. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X. Effects of fat on glucose uptake and utilization in patients with non-insulin-dependent diabetes. J. Clin. Invest. 1995, 96, 1261–1268. [Google Scholar]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 1996, 97, 2859–2865. [Google Scholar] [CrossRef]
- Rebrin, K.; Steil, G.M.; Getty, L.; Bergman, R.N. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes 1995, 44, 1038–1045. [Google Scholar]
- Boden, G.; Jadali, F.; White, J.; Liang, Y.; Mozzoli, M.; Chen, X.; Coleman, E.; Smith, C. Effects of fat on insulin-stimulated carbohydrate metabolism in normal men. J. Clin. Invest. 1991, 88, 960–966. [Google Scholar] [CrossRef]
- Hennes, M.M.; Shrago, E.; Kissebah, A.H. Receptor and postreceptor effects of free fatty acids (FFA) on hepatocyte insulin dynamics. Int. J. Obes. 1990, 14, 831–841. [Google Scholar]
- Kissebah, A.H.; Alfarsi, S.; Adams, P.W.; Wynn, V. Role of insulin resistance in adipose tissue and liver in the pathogenesis of endogenous hypertriglyceridaemia in man. Diabetologia 1976, 12, 563–571. [Google Scholar] [CrossRef]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J.M. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Invest. 1989, 83, 1168–1173. [Google Scholar] [CrossRef]
- Guo, Z.; Hensrud, D.D.; Johnson, C.M.; Jensen, M.D. Regional postprandial fatty acid metabolism in different obesity phenotypes. Diabetes 1999, 48, 1586–1592. [Google Scholar] [CrossRef]
- Roust, L.R.; Jensen, M.D. Postprandial free fatty acid kinetics are abnormal in upper body obesity. Diabetes 1993, 42, 1567–1573. [Google Scholar]
- Basso, L.V.; Havel, R.J. Hepatic metabolism of free fatty acids in normal and diabetic dogs. J. Clin. Invest. 1970, 49, 537–547. [Google Scholar] [CrossRef]
- Jensen, M.D.; Cardin, S.; Edgerton, D.; Cherrington, A. Splanchnic free fatty acid kinetics. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1140–E1148. [Google Scholar]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Invest. 2004, 113, 1582–1588. [Google Scholar]
- Kissebah, A.H.; Adams, P.W.; Wynn, V. Plasma free fatty acid and triglyceride transport kinetics in man. Clin. Sci. Mol. Med. 1974, 47, 259–278. [Google Scholar]
- McBride, P. Triglycerides and risk for coronary artery disease. Curr. Atheroscler. Rep. 2008, 10, 386–390. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.F.; Hokanson, J.E. The complex role of triglycerides in cardiovascular disease. Semin. Vasc. Med. 2002, 2, 325–333. [Google Scholar] [CrossRef]
- Veilleux, A.; Caron-Jobin, M.; Noel, S.; Laberge, P.Y.; Tchernof, A. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes 2011, 60, 1504–1511. [Google Scholar] [CrossRef]
- Lemieux, I.; Poirier, P.; Bergeron, J.; Almeras, N.; Lamarche, B.; Cantin, B.; Dagenais, G.R.; Despres, J.P. Hypertriglyceridemic waist: a useful screening phenotype in preventive cardiology? Can. J. Cardiol. 2007, 23, 23B–31B. [Google Scholar] [CrossRef]
- Choi, S.H.; Ginsberg, H.N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol. Metab. 2011, 22, 353–363. [Google Scholar] [CrossRef]
- Rashid, S.; Uffelman, K.D.; Lewis, G.F. The mechanism of HDL lowering in hypertriglyceridemic, insulin-resistant states. J. Diabetes Complications 2002, 16, 24–28. [Google Scholar]
- Despres, J.P.; Moorjani, S.; Tremblay, A.; Ferland, M.; Lupien, P.J.; Nadeau, A.; Bouchard, C. Relation of high plasma triglyceride levels associated with obesity and regional adipose tissue distribution to plasma lipoprotein-lipid composition in premenopausal women. Clin. Invest. Med. 1989, 12, 374–380. [Google Scholar]
- Eisenberg, S. Lipoprotein abnormalities in hypertriglyceridemia: Significance in atherosclerosis. Am. Heart J. 1987, 113, 555–561. [Google Scholar] [CrossRef]
- Meek, S.E.; Nair, K.S.; Jensen, M.D. Insulin regulation of regional free fatty acid metabolism. Diabetes 1999, 48, 10–14. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar]
- Basu, A.; Basu, R.; Shah, P.; Vella, A.; Rizza, R.A.; Jensen, M.D. Systemic and regional free fatty acid metabolism in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E1000–E1006. [Google Scholar]
- Cohn, J.S.; Patterson, B.W.; Uffelman, K.D.; Davignon, J.; Steiner, G. Rate of production of plasma and very-low-density lipoprotein (VLDL) apolipoprotein C-III is strongly related to the concentration and level of production of VLDL triglyceride in male subjects with different body weights and levels of insulin sensitivity. J. Clin. Endocrinol. Metab. 2004, 89, 3949–3955. [Google Scholar] [CrossRef]
- Krause, B.R.; Hartman, A.D. Adipose tissue and cholesterol metabolism. J. Lipid Res. 1984, 25, 97–110. [Google Scholar]
- Wang, H.; Peng, D.Q. New insights into the mechanism of low high-density lipoprotein cholesterol in obesity. Lipids Health Dis. 2011, 10, 176. [Google Scholar] [CrossRef]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Castelli, W.P.; Garrison, R.J.; Wilson, P.W.; Abbott, R.D.; Kalousdian, S.; Kannel, W.B. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA 1986, 256, 2835–2838. [Google Scholar] [CrossRef]
- Gotto, A.M., Jr.; Whitney, E.; Stein, E.A.; Shapiro, D.R.; Clearfield, M.; Weis, S.; Jou, J.Y.; Langendorfer, A.; Beere, P.A.; Watson, D.J.; et al. Relation between baseline and on-treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Circulation 2000, 101, 477–484. [Google Scholar]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R., Jr.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef]
- Shah, P.K.; Kaul, S.; Nilsson, J.; Cercek, B. Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteins: An idea whose time for testing is coming, part I. Circulation 2001, 104, 2376–2383. [Google Scholar] [CrossRef]
- Kekki, M. Lipoprotein-lipase action determining plasma high density lipoprotein cholesterol level in adult normolipaemics. Atherosclerosis 1980, 37, 143–150. [Google Scholar] [CrossRef]
- Eisenberg, S. Lipoproteins and lipoprotein metabolism. A dynamic evaluation of the plasma fat transport system. Klin. Wochenschr. 1983, 61, 119–132. [Google Scholar] [CrossRef]
- Eisenberg, S. High density lipoprotein metabolism. J. Lipid Res. 1984, 25, 1017–1058. [Google Scholar]
- Nikkila, E.A.; Taskinen, M.R.; Sane, T. Plasma high-density lipoprotein concentration and subfraction distribution in relation to triglyceride metabolism. Am. Heart J. 1987, 113, 543–548. [Google Scholar]
- Tikkanen, M.J.; Nikkila, E.A. Regulation of hepatic lipase and serum lipoproteins by sex steroids. Am. Heart J. 1987, 113, 562–567. [Google Scholar] [CrossRef]
- Pouliot, M.C.; Despres, J.P.; Moorjani, S.; Lupien, P.J.; Tremblay, A.; Nadeau, A.; Bouchard, C. Regional variation in adipose tissue lipoprotein lipase activity: Association with plasma high density lipoprotein levels. Eur. J. Clin. Invest. 1991, 21, 398–405. [Google Scholar] [CrossRef]
- Despres, J.P.; Ferland, M.; Moorjani, S.; Nadeau, A.; Tremblay, A.; Lupien, P.J.; Theriault, G.; Bouchard, C. Role of hepatic-triglyceride lipase activity in the association between intra-abdominal fat and plasma HDL cholesterol in obese women. Arteriosclerosis 1989, 9, 485–492. [Google Scholar] [CrossRef]
- Carr, M.C.; Hokanson, J.E.; Zambon, A.; Deeb, S.S.; Barrett, P.H.; Purnell, J.Q.; Brunzell, J.D. The contribution of intraabdominal fat to gender differences in hepatic lipase activity and low/high density lipoprotein heterogeneity. J. Clin. Endocrinol. Metab. 2001, 86, 2831–2837. [Google Scholar] [CrossRef]
- Guendouzi, K.; Jaspard, B.; Barbaras, R.; Motta, C.; Vieu, C.; Marcel, Y.; Chap, H.; Perret, B.; Collet, X. Biochemical and physical properties of remnant-HDL2 and of pre beta 1-HDL produced by hepatic lipase. Biochemistry 1999, 38, 2762–2768. [Google Scholar] [CrossRef]
- De Souza, J.A.; Vindis, C.; Hansel, B.; Negre-Salvayre, A.; Therond, P.; Serrano, C.V., Jr.; Chantepie, S.; Salvayre, R.; Bruckert, E.; Chapman, M.J.; Kontush, A. Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis 2008, 197, 84–94. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ebbert, J.O.; Jensen, M.D. Fat Depots, Free Fatty Acids, and Dyslipidemia. Nutrients 2013, 5, 498-508. https://doi.org/10.3390/nu5020498
Ebbert JO, Jensen MD. Fat Depots, Free Fatty Acids, and Dyslipidemia. Nutrients. 2013; 5(2):498-508. https://doi.org/10.3390/nu5020498
Chicago/Turabian StyleEbbert, Jon O., and Michael D. Jensen. 2013. "Fat Depots, Free Fatty Acids, and Dyslipidemia" Nutrients 5, no. 2: 498-508. https://doi.org/10.3390/nu5020498
APA StyleEbbert, J. O., & Jensen, M. D. (2013). Fat Depots, Free Fatty Acids, and Dyslipidemia. Nutrients, 5(2), 498-508. https://doi.org/10.3390/nu5020498