The Use of Fish Oil Lipid Emulsion in the Treatment of Intestinal Failure Associated Liver Disease (IFALD)

Abstract
:1. Introduction
1.1. Essential Fatty Acids (EFAs)
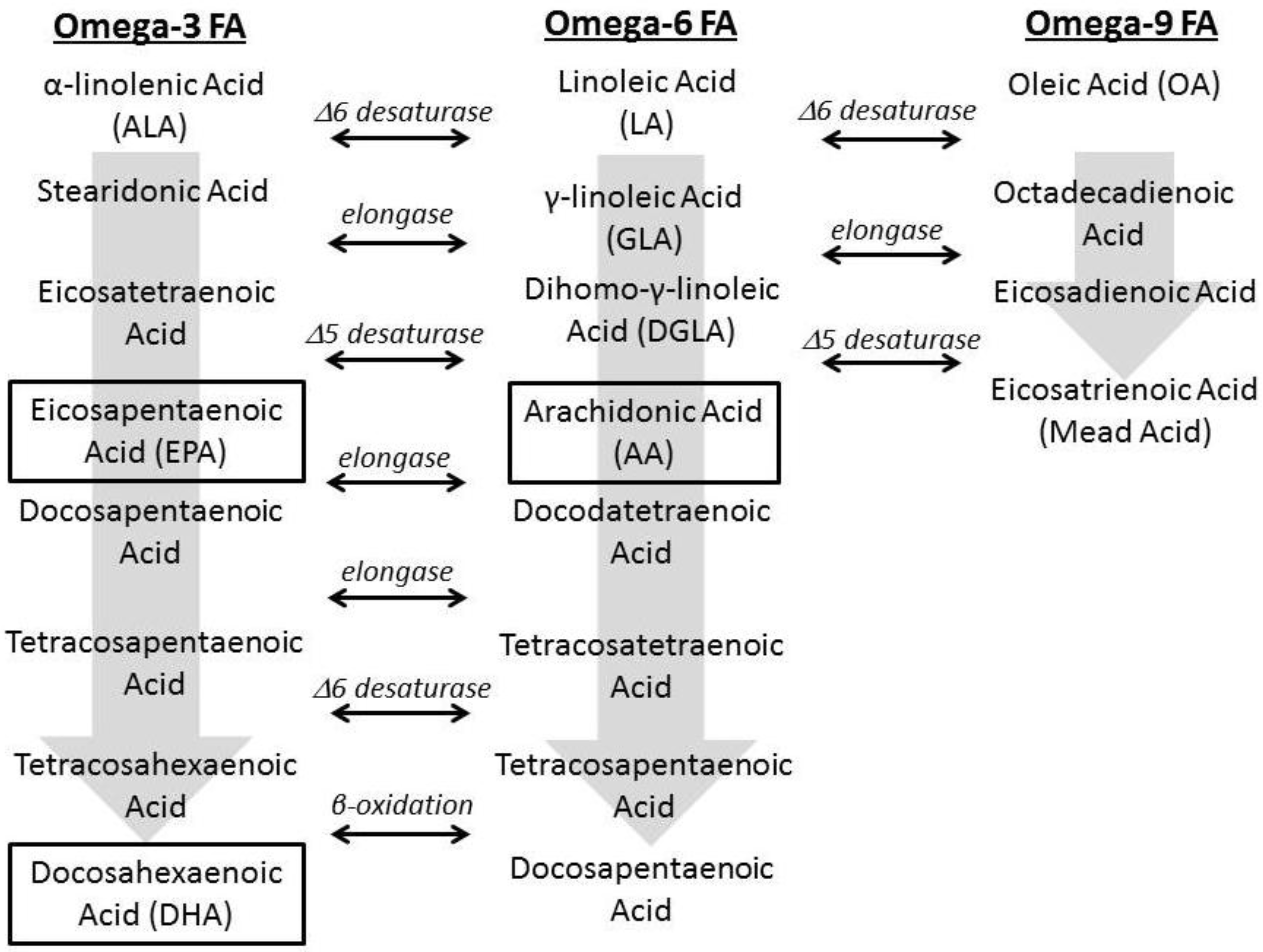
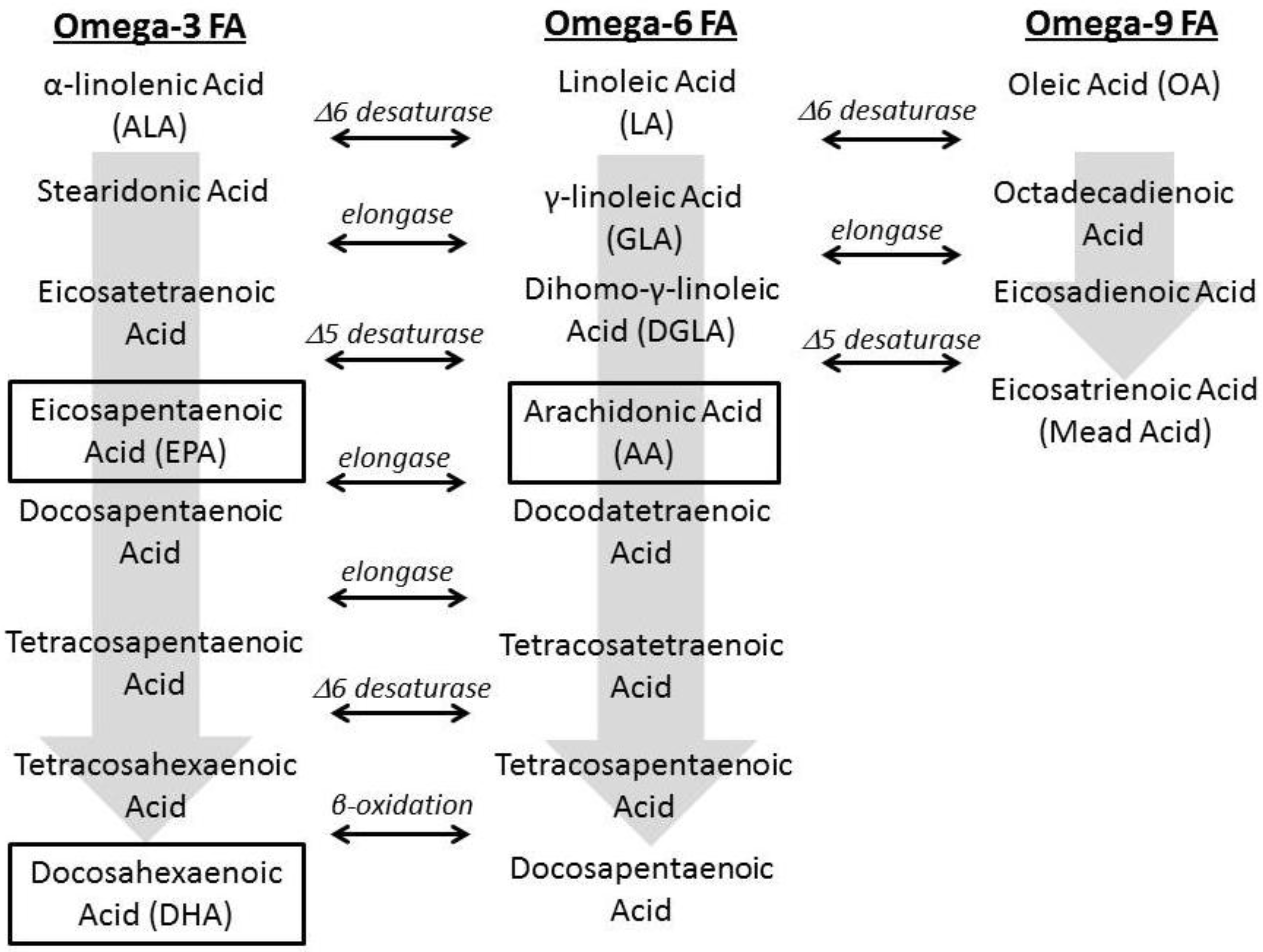
1.2. Fatty Acid Regulation
1.3. Role of Fatty Acids in Inflammation
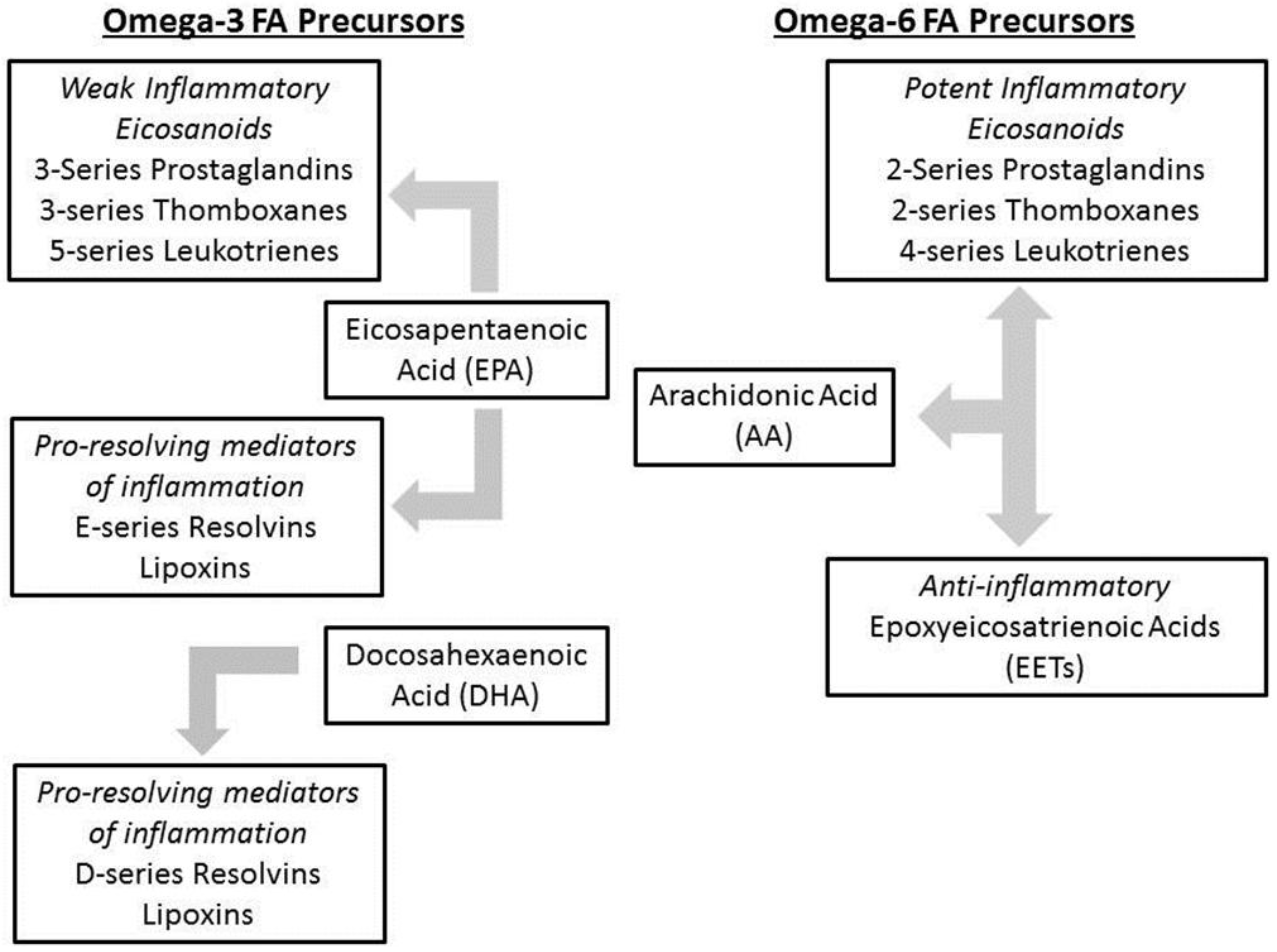
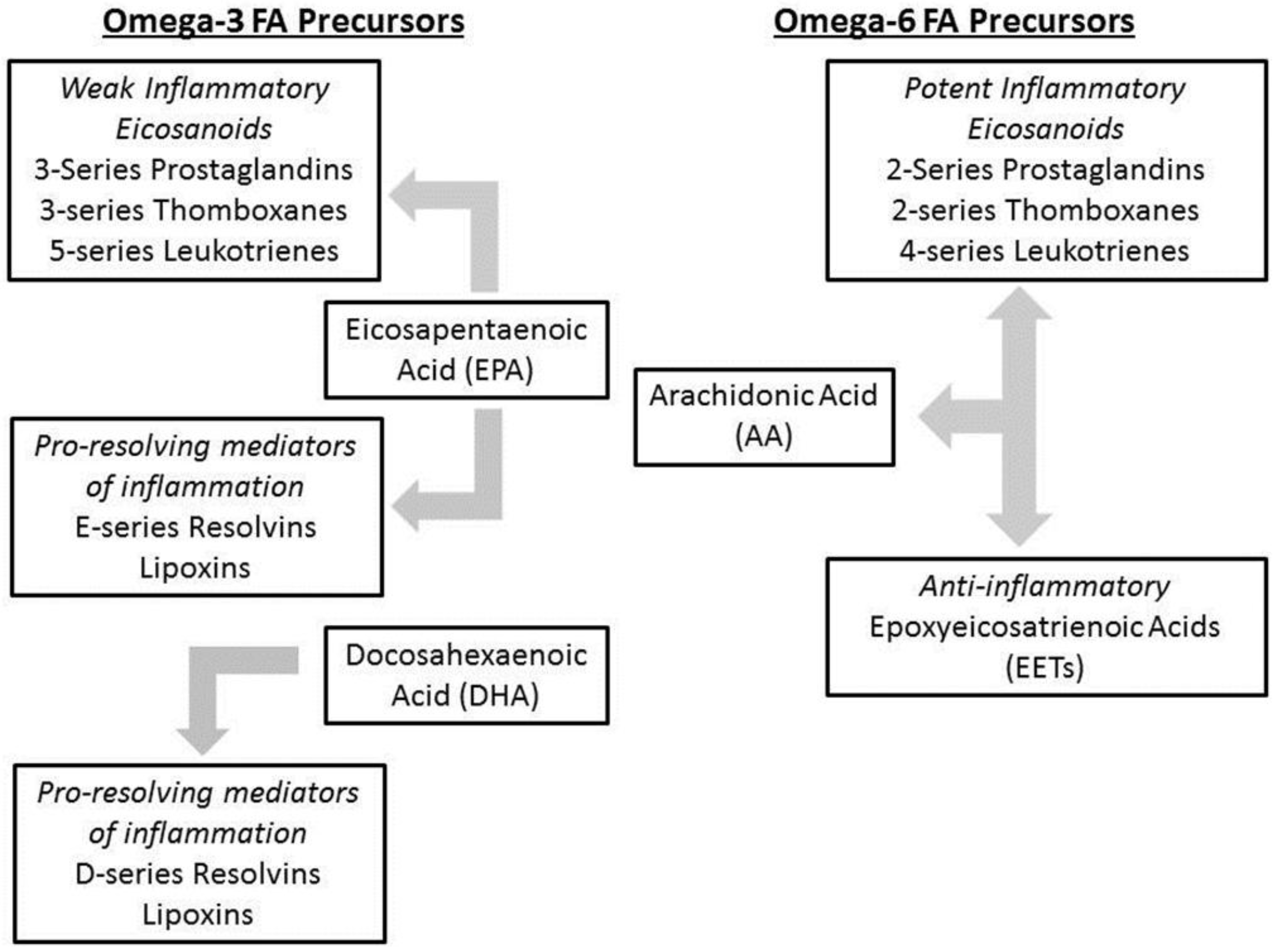
1.4. Intestinal Failure Associated Liver Disease (IFALD)
1.5. Lipid Emulsions and IFALD

{kind=link}
{kind=link}
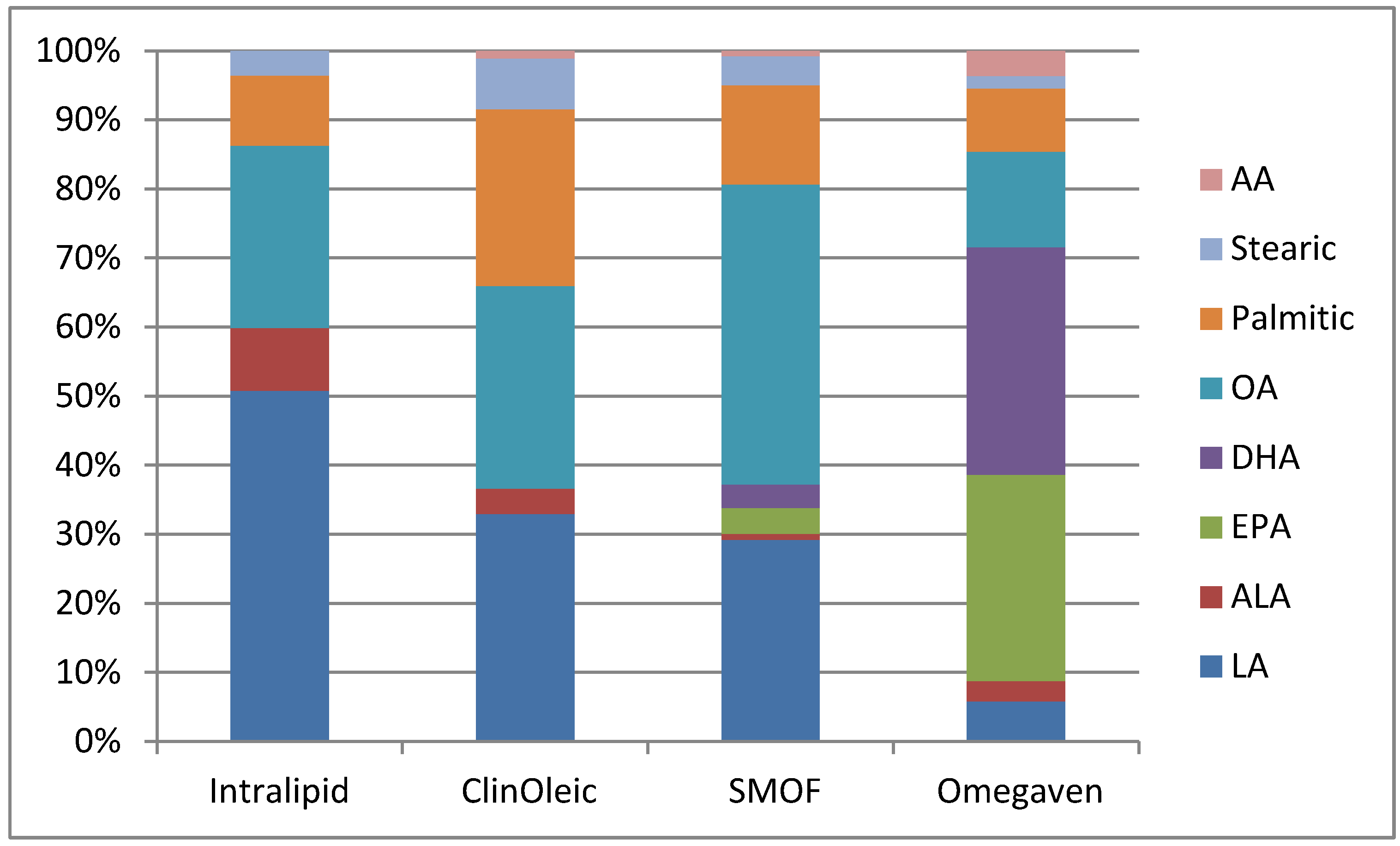
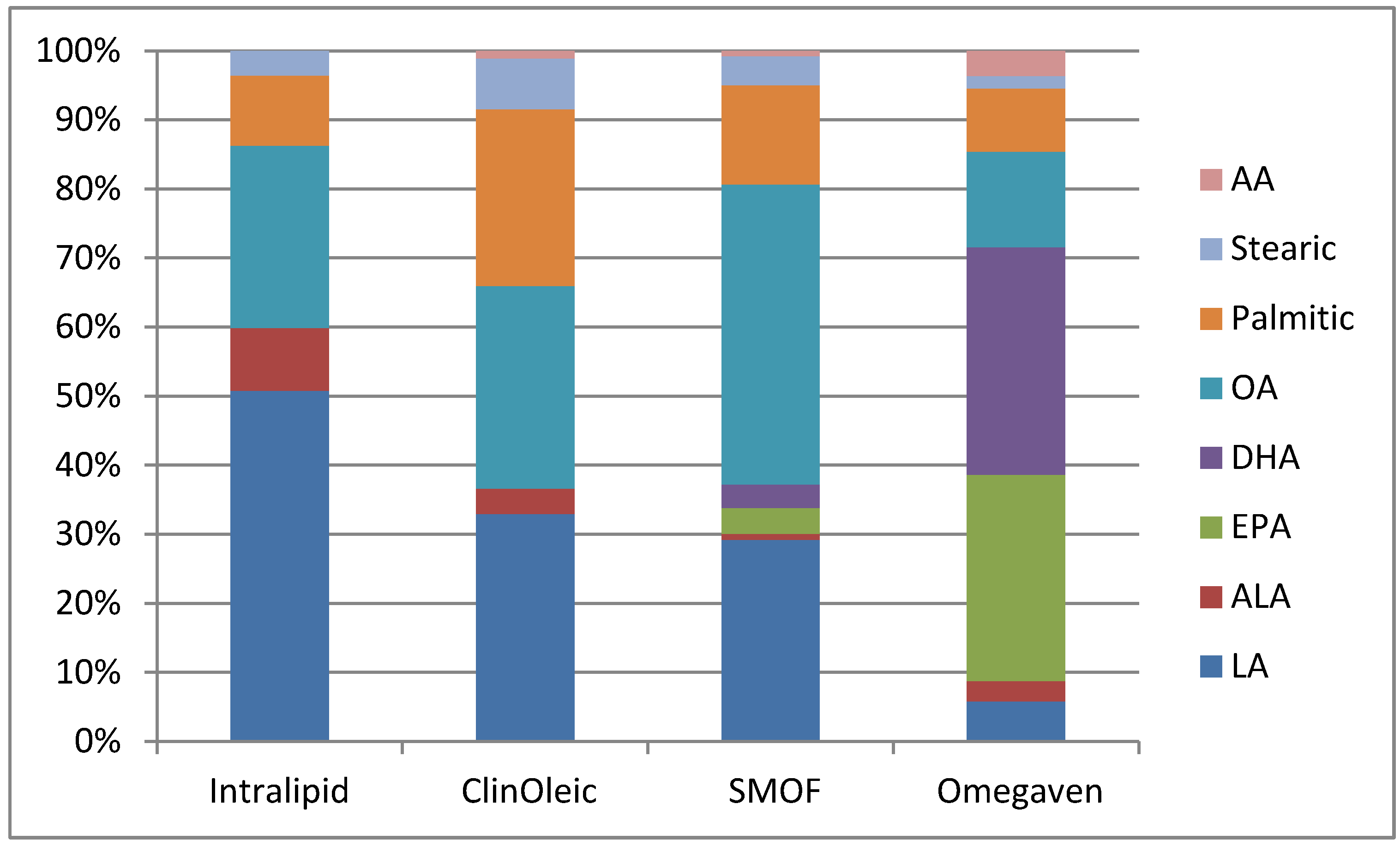{kind=link}
| Product | Intralipid | ClinOleic | SMOF lipid | Omegaven |
|---|---|---|---|---|
| Manufacturer | Baxter Healthcare/Fresenius Kabi | Baxter Healthcare/Parenteral SA | Fresenius Kabi | Fresenius Kabi |
| Oil source (g) | ||||
| Soy bean | 10 | 2 | 3 | 0 |
| Safflower | 0 | 0 | 0 | 0 |
| MCT | 0 | 0 | 3 | 0 |
| Olive oil | 0 | 8 | 2.5 | 0 |
| Fish oil | 0 | 0 | 1.5 | 10 |
| Alpha Tocopherol | 38 | 32 | 200 | 150–296 |
| Phytosterols (mg/L) | 348 ± 33 | 327 ± 8 | 47.6 | 0 |
| Fat composition (g) | ||||
| Linoleic | 5 | 0.9 | 1.87 | 0.1–0.7 |
| Alpha-linolenic | 0.9 | 0.1 | 0.24 | <0.2 |
| EPA | 0 | 0 | 0.24 | 1.28–2.82 |
| DHA | 0 | 0 | 0.22 | 1.44–3.09 |
| Oleic | 2.6 | 0.8 | 2.78 | 0.6–1.3 |
| Palmitic | 1 | 0.7 | 0.92 | 0.25–1 |
| Stearic | 0.35 | 0.2 | 0.27 | 0.05–0.2 |
| Arachidonic | 0 | 0.03 | 0.05 | 0.1–0.4 |
1.6. Clinical Experience
1.7. Alternative IVLE Dosage Strategies
2. Conclusions
Conflict of Interest
References
- Dudrick, S.J.; Wilmore, D.W.; Vars, H.M.; Rhoads, J.E. Long-term total parenteral nutrition with growth, development, and positive nitrogen balance. Surgery 1968, 64, 134–142. [Google Scholar]
- Squires, R.H.; Duggan, C.; Teitelbaum, D.H.; Wales, P.W.; Balint, J.; Venick, R.; Rhee, S.; Sudan, D.; Mercer, D.; Martinez, J.A.; et al. Natural history of pediatric intestinal failure: Initial report from the pediatric intestinal failure consortium. J. Pediatr. 2012, 161, 723–728. [Google Scholar] [CrossRef]
- Quiros-Tejeira, R.E.; Ament, M.E.; Reyen, L.; Herzog, F.; Merjanian, M.; Olivares-Serrano, N.; Vargas, J.H. Long-term parenteral nutritional support and intestinal adaptation in children with short bowel syndrome: A 25-year experience. J. Pediatr. 2004, 145, 157–163. [Google Scholar] [CrossRef]
- Willis, T.C.; Carter, B.A.; Rogers, S.P.; Hawthorne, K.M.; Hicks, P.D.; Abrams, S.A. High rates of mortality and morbidity occur in infants with parenteral nutrition-associated cholestasis. JPEN J. Parenter. Enteral Nutr. 2010, 34, 32–37. [Google Scholar] [CrossRef]
- Chan, S.; McCowen, K.C.; Bistrian, B.R.; Thibault, A.; Keane-Ellison, M.; Forse, R.A.; Babineau, T.; Burke, P. Incidence, prognosis, and etiology of end-stage liver disease in patients receiving home total parenteral nutrition. Surgery 1999, 126, 28–34. [Google Scholar] [CrossRef]
- Diamanti, A.; Basso, M.S.; Castro, M.; Calce, A.; Pietrobattista, A.; Gambarara, M. Prevalence of life-threatening complications in pediatric patients affected by intestinal failure. Transplant. Proc. 2007, 39, 1632–1633. [Google Scholar] [CrossRef]
- Andorsky, D.J.; Lund, D.P.; Lillehei, C.W.; Jaksic, T.; Dicanzio, J.; Richardson, D.S.; Collier, S.B.; Lo, C.; Duggan, C. Nutritional and other postoperative management of neonates with short bowel syndrome correlates with clinical outcomes. J. Pediatr. 2001, 139, 27–33. [Google Scholar] [CrossRef]
- Beath, S.V.; Davies, P.; Papadopoulou, A.; Khan, A.R.; Buick, R.G.; Corkery, J.J.; Gornall, P.; Booth, I.W. Parenteral nutrition-related cholestasis in postsurgical neonates: Multivariate analysis of risk factors. J. Pediatr. Surg. 1996, 31, 604–606. [Google Scholar] [CrossRef]
- Christensen, R.D.; Henry, E.; Wiedmeier, S.E.; Burnett, J.; Lambert, D.K. Identifying patients, on the first day of life, at high-risk of developing parenteral nutrition-associated liver disease. J. Perinatol. 2007, 27, 284–290. [Google Scholar]
- Burr, G.O.; Burr, M.M. A new deficiency disease produced by the rigid exclusion of fat from the diet. J. Biol. Chem. 1929, 82, 230–234. [Google Scholar]
- Holman, R.T. The ratio of trienoic: Tetraenoic acids in tissue lipids as a measure of essential fatty acid requirement. J. Nutr. 1960, 70, 405–410. [Google Scholar]
- Connor, W.E. Pathogenesis and frequency of essential fatty acid deficiency during total parenteral nutrition. Ann. Intern. Med. 1975, 83, 895–896. [Google Scholar]
- Tashiro, T.; Ogata, H.; Yokoyama, H.; Mashima, Y.; Iwasaki, I. The effect of fat emulsion of essential fatty acid deficiency during intravenous hyperalimentation in pediatric patients. J. Pediatr. Surg. 1975, 10, 203–213. [Google Scholar] [CrossRef]
- Friedman, A.; Moe, S. Review of the effects of omega-3 supplementation in dialysis patients. Clin. J. Am. Soc. Nephrol. 2006, 1, 182–192. [Google Scholar] [CrossRef]
- Smit, E.N.; Muskiet, F.A.; Boersma, E.R. The possible role of essential fatty acids in the pathophysiology of malnutrition: A review. Prostaglandins Leukot. Essent. Fatty Acids 2004, 71, 241–250. [Google Scholar]
- Holman, R. Essential fatty acid deficiency. Prog. Chem. Fats Other Lipids 1971, 9, 275–348. [Google Scholar] [CrossRef]
- Mohrhauer, H.; Holman, R.T. The effect of dose level of essential fatty acids upon fatty acid composition of the rat liver. J. Lipid Res. 1963, 4, 151–159. [Google Scholar]
- Clarke, S.D.; Jump, D.B. Dietary polyunsaturated fatty acid regulation of gene transcription. Annu. Rev. Nutr. 1994, 14, 83–98. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 2004, 114, 147–152. [Google Scholar]
- Jump, D.B. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr. Opin. Lipidol. 2008, 19, 242–247. [Google Scholar] [CrossRef]
- Wang, Y.; Botolin, D.; Christian, B.; Busik, J.; Xu, J.; Jump, D.B. Tissue-specific, nutritional, and developmental regulation of rat fatty acid elongases. J. Lipid Res. 2005, 26, 706–715. [Google Scholar]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; Kliewer, S.A.; Milburn, M.V. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Fibroblast growth factor 21: From pharmacology to physiology. Am. J. Clin. Nutr. 2010, 91, 254S–257S. [Google Scholar] [CrossRef]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar]
- Park, Y.; Harris, W. Omega-3 fatty acid supplementation accelerates chylomicron triglyceride clearance. J. Lipid Res. 2003, 44, 455–463. [Google Scholar] [CrossRef]
- Fisher, E.A.; Williams, K.J. Autophagy of an oxidized, aggregated protein beyond the ER: A pathway for remarkably late-stage quality control. Autophagy 2008, 4, 721–723. [Google Scholar]
- Brodsky, J.L.; Fisher, E.A. The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol. Metab. 2008, 19, 254–259. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Fisher, E.A. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J. Lipid Res. 2009, 50, S162–S166. [Google Scholar] [CrossRef]
- Jump, D.B.; Botolin, D.; Wang, Y.; Xu, J.; Demeure, O.; Christian, B. Docosahexaenoic acid (DHA) and hepatic gene transcription. Chem. Phys. Lipids 2008, 153, 3–13. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty acids: Biochemistry, physiology, and pathology. Biotechnol. J. 2006, 1, 420–439. [Google Scholar]
- Dichtl, W.; Ares, M.P.; Jonson, A.N.; Jovinge, S.; Pachinger, O.; Giachelli, C.M.; Hamsten, A.; Eriksson, P.; Nilsson, J. Linoleic acid-stumulated vascular adhesion molecule-1 expression in endothelial cells depends on nuclear factor-kappa B activation. Metabolism 2002, 51, 327–333. [Google Scholar]
- Park, H.J.; Lee, Y.W.; Hennig, B.; Toberek, M. Linoleic acid-induced VCAM-1 expression in human microvascular endothelial cells is mediated by the NF-kappa B-dependent pathway. Nutr. Cancer 2001, 41, 126–134. [Google Scholar]
- Calder, P. Immunomodulation by omega-3 fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2007, 77, 327–335. [Google Scholar] [CrossRef]
- Wall, R.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef]
- Sijben, J.W.C.; Calder, P.C. Differential immunomodulation with long chain n-3 PUFA in health and chronic disease. Proc. Nutr. Soc. 2007, 66, 237–259. [Google Scholar]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Klaus, L.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory Properties of Cytochrome P450 Epoxygenase-Derived Eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef]
- Spector, A.A.; Fang, X.; Snyder, G.D.; Weintraub, N.L. Epoxyeicosatrienoic Acids (EETs): Metabolism and Biochemical Function. Prog. Lipid Res. 2004, 43, 55–90. [Google Scholar] [CrossRef]
- Cleland, L.G.; James, M.J.; Proudman, S.M. The role of fish oils in the treatment of rheumatoid arthritis. Drugs 2003, 63, 845–853. [Google Scholar] [CrossRef]
- Bagga, D.; Wang, L.; Farias-Eisner, R.; Glaspy, J.A.; Reddy, S.T. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 1751–1756. [Google Scholar]
- Bellenger, J.; Bellenger, S.; Clement, L.; Mandard, S.; Diot, C.; Poisson, J.P.; Narce, M. A new hypotensive polyunsaturated fatty acid dietary combination regulates oleic acid accumulation by suppression of stearoyl CoA desaturase gene 1 gene expression in the SHR model of genetic hypertension. FASEB J. 2004, 18, 773–775. [Google Scholar]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-Inflammatory and Insulin Sensitizing Effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef]
- Zhao, Y.; Joshi-Barve, S.; Barve, S.; Chen, L.H. Eicosapentaenoic acid prevents LPS-induced TNF-α expression by preventing NF-kB activation. J. Am. Coll. Nutr. 2004, 23, 71–78. [Google Scholar]
- Calder, P.C. Mechanisms of Action of (n-3) Fatty Acids. J. Nutr. 2012, 142, 592–599. [Google Scholar] [CrossRef]
- Leaf, A.; Weber, P.C. Cardiovascular effects of n-3 fatty acids. N. Engl. J. Med. 1988, 318, 549–557. [Google Scholar] [CrossRef]
- Schmidt, E.B.; Dyerberg, J. Omega-3 fatty acids. Current status in cardiovascular medicine. Drugs 1994, 47, 405–424. [Google Scholar] [CrossRef]
- Grimminger, F.; Wahn, H.; Mayer, K.; Kiss, L.; Walmrath, D.; Seeger, W. Impact of arachidonic versus eicosapentaenoic acid on exotonin-induced lung vascular leakage: Relation to 4-series versus 5-series leukotriene generation. Am. J. Respir. Crit. Care Med. 1997, 155, 513–519. [Google Scholar]
- Lee, T.H.; Hoover, R.L.; Williams, J.D.; Sperling, R.I.; Ravalese, J., III; Spur, B.W.; Robinson, D.R.; Corey, E.J.; Lewis, R.A.; Austen, K.F. Effect of dietary enrichment with eicosapentaenoic and docosahexaenoic acids on in vitro neutrophil and monocyte leukotriene generation and neutrophil function. N. Engl. J. Med. 1985, 312, 1217–1224. [Google Scholar]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 8, 1025–1037. [Google Scholar]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar]
- Connor, K.M.; SanGiovanni, J.P.; Lofqvist, C.; Aderman, C.M.; Chen, J.; Higuchi, A.; Hong, S.; Pravda, E.A.; Majchrzak, S.; Carper, D.; et al. Increased dietary intake of omega-3 polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 2007, 13, 868–873. [Google Scholar]
- Seki, H.; Fukunaga, K.; Arita, M.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; Takeda, J.; Levy, B.D. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar]
- Duffield, J.S.; Hong, S.; Vaidya, V.S.; Lu, Y.; Fredman, G.; Serhan, C.N.; Bonventre, J.V. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 2006, 177, 5902–5911. [Google Scholar]
- Recchiuti, A.; Krishnamoorthy, S.; Fredman, G.; Chiang, N.; Serhan, C.N. MicroRNAs in resolution of acute inflammation: Identification of novel resolvin D1-miRNA circuits. FASEB J. 2011, 25, 544–560. [Google Scholar]
- Xu, Z.Z.; Zhang, L.; Liu, T.; Park, J.Y.; Berta, T.; Yang, R.; Serhan, C.N.; Ji, R.R. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 2010, 16, 592–597. [Google Scholar]
- Gonzalez-Periz, A.; Horrillo, R.; Ferre, N.; Gronert, K.; Dong, B.; Moran-Salvador, E.; Titos, E.; Martinez-Clemente, M.; Lopez-Parra, M.; Arroyo, V.; Claria, J. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 6, 1946–1957. [Google Scholar]
- Vanek, V.W.; Seidner, D.L.; Allen, P.; Bistrian, B.; Collier, S.; Gura, K.; Miles, J.M.; Valentine, C.J.; Kochevar, M. Novel Nutrient Task Force, Intravenous Fat Emulsions Workgroup; American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.) Board of Directors. A.S.P.E.N. position paper: Clinical role for alternative intravenous fat emulsions. Nutr. Clin. Pract. 2012, 27, 150–192. [Google Scholar]
- Wanten, G.J.; Calder, P.C. Immune modulation by parenteral lipid emulsions. Am. J. Clin. Nutr. 2007, 85, 1171–1184. [Google Scholar]
- Seidner, D.L.; Mascioli, E.A.; Istfan, N.W.; Porter, K.A.; Selleck, K.; Blackburn, G.L.; Bistrian, B.R. Effects of long chain triglyceride emulsions on reticuloendothelial system function in humans. JPEN J. Parenter. Enteral Nutr. 1989, 13, 614–619. [Google Scholar]
- Battistella, F.D.; Widergren, J.T.; Anderson, J.T.; Siepler, J.K.; Weber, J.C.; MacColl, K. A prospective, randomized trial of intravenous fat emulsion administration in trauma victims requiring total parenteral nutrition. J. Trauma 1997, 43, 52–60. [Google Scholar] [CrossRef]
- Cober, M.P.; Teitelbaum, D.H. Prevention of parenteral nutrition-associated liver disease: Lipid minimization. Curr. Opin. Organ.Transplant. 2010, 15, 330–333. [Google Scholar] [CrossRef]
- Meisel, J.A.; Le, H.D.; de Meijer, V.E.; Nose, V.; Gura, K.M.; Mulkern, R.V.; Akhavan Sharif, M.R.; Puder, M. Comparison of 5 intravenous lipid emulsions and their effects on hepatic steatosis in a murine model. J. Pediatr. Surg. 2011, 46, 666–673. [Google Scholar]
- Driscoll, D.F. Lipid injectable emulsions: 2006. Nutr. Clin. Pract. 2006, 21, 381–386. [Google Scholar] [CrossRef]
- Freund, H.R. Abnormalities of liver function and hepatic damage associated with total parenteral nutrition. Nutrition 1991, 7, 1–6. [Google Scholar]
- Greenberg, G.R.; Wolman, S.L.; Christofides, N.D.; Bloom, S.R.; Jeejeebhoy, K.N. Effect of total parenteral nutrition on gut hormone release in humans. Gastroenterology 1981, 80, 988–993. [Google Scholar]
- Yeh, S.L.; Chen, W.J.; Huang, P.C. Effects of L-glutamine on induced hepatosteatosis in rats receiving total parenteral nutrition. J. Formos. Med. Assoc. 1995, 94, 593–599. [Google Scholar]
- Kubota, A.; Yonekura, T.; Hoki, M.; Oyanagi, H.; Kawahara, H.; Yagi, M.; Imura, K.; Iiboshi, Y.; Wasa, K.; Kamata, S.; Okada, A. Total parenteral nutrition-associated intrahepatic cholestasis in infants: 25 years’ experience. J. Pediatr. Surg. 2000, 35, 1049–1051. [Google Scholar] [CrossRef]
- Moss, R.L.; Das, J.B.; Ansari, G.; Raffensperger, J.G. Hepatobiliary dysfunction during total parenteral nutrition is caused by infusate, not the route of administration. J. Pediatr. Surg. 1993, 28, 391–397. [Google Scholar]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Kaminski, D.L.; Adams, A.; Jellinek, M. The effect of hyperalimentation on hepatic lipid content and lipogenic enzyme activity in rats and man. Surgery 1980, 88, 93–100. [Google Scholar]
- Hultin, M.; Carneheim, C.; Rosenqvist, K.; Olivecrona, T. Intravenous lipid emulsions: Removal mechanisms as compared to chylomicrons. J. Lipid Res. 1995, 36, 2174–2184. [Google Scholar]
- Li, S.J.; Nussbaum, M.S.; McFadden, D.W.; Dayal, R.; Fischer, J.E. Reversal of hepatic steatosis in rats by addition of glucagon to total parenteral nutrition (TPN). J. Surg. Res. 1989, 46, 557–566. [Google Scholar] [CrossRef]
- Rangel, S.J.; Calkins, C.M.; Cowles, R.A.; Barnhart, D.C.; Huang, E.Y.; Abdullah, F.; Arca, M.J.; Teitelbaum, D.H. 2011 American Pediatric Surgical Association Outcomes and Clinical Trials Committee. Parenteral nutrition-associated cholestasis: An American Pediatric Surgical Association Outcomes and Clinical Trials Committee systematic review. J. Pediatr. Surg. 2012, 47, 225–240. [Google Scholar]
- Qi, K.; Al-Haideri, M.; Seo, T.; Carpentier, Y.A.; Deckelbaum, R.J. Effects of particle size on blood clearance and tissue uptake of lipid emulsions with different triglyceride compositions. JPEN J. Parenter. Enteral Nutr. 2003, 27, 58–64. [Google Scholar]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef]
- Ruan, H.; Lodish, H.F. Insulin resistance in adipose tissue: Direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003, 14, 447–455. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef]
- Li, H.; Miyahara, T.; Tezuka, Y.; Tran, Q.L.; Seto, H.; Kadota, S. Effect of berberine on bone mineral density in SAMP6 as a senile osteoporosis model. Biol. Pharm. Bull. 2003, 26, 110–111. [Google Scholar]
- Feingold, K.R.; Soued, M.; Grunfeld, C. Tumor necrosis factor stimulates DNA synthesis in the liver of intact rats. Biochem. Biophys. Res. Commun. 1988, 153, 576–582. [Google Scholar]
- Feldstein, A.E.; Canbay, A.; Guicciardi, M.E.; Higuchi, H.; Bronk, S.F.; Gores, G.J. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J. Hepatol. 2003, 39, 978–983. [Google Scholar] [CrossRef]
- Mullick, F.G.; Moran, C.A.; Ishak, K.G. Total parenteral nutrition: A histopathologic analysis of the liver changes in 20 children. Mod. Pathol. 1994, 7, 190–194. [Google Scholar]
- Kaufman, S.S.; Pehlivanova, M.; Fennelly, E.M.; Rekhtman, Y.M.; Gondolesi, G.E.; Little, C.A.; Matsumoto, C.S.; Fishbein, T.M. Predicting liver failure in parenteral nutrition-dependent short bowel syndrome of infancy. J. Pediatr. 2010, 156, 580–585.e1. [Google Scholar] [CrossRef]
- Nehra, D.; Fallon, E.M.; Puder, M. The prevention and treatment of intestinal failure-associated liver disease in neonates and children. Surg. Clin. North Am. 2011, 91, 543–563. [Google Scholar]
- Goulet, O.; Joly, F.; Corriol, O.; Colomb-Jung, V. Some new insights in intestinal failure-associated liver disease. Curr. Opin. Organ. Transplant. 2009, 14, 256–261. [Google Scholar]
- Keim, N.L.; Mares-Perlman, J.A. Development of hepatic steatosis and essential fatty acid deficiency in rats with hypercaloric, fat-free parenteral nutrition. J. Nutr. 1984, 114, 1807–1815. [Google Scholar]
- Goheen, S.C.; Larkin, E.C.; Rao, G.A. Severe fatty liver in rats fed a fat-free ethanol diet, and its prevention by small amounts of dietary arachidonate. Lipids 1983, 18, 285–290. [Google Scholar] [CrossRef]
- Javid, P.; Greene, A.; Garza, J.; Gura, K.; Alwayn, I.P.; Voss, S.; Nose, V.; Satchi-Fainaro, R.; Zausche, B.; Mulkern, R.V.; Jaksic, T.; Bistrian, B.; Folkman, J.; Puder, M. The route of lipid administration affects parenteral nutrition-induced hepatic steatosis in a mouse model. J. Pediatr. Surg. 2005, 40, 1446–1453. [Google Scholar] [CrossRef]
- Xu, Z.; Harvey, K.A.; Pavlina, T.; Dutot, G.; Hise, M.; Zaloga, G.P.; Siddiqui, R.A. Steroidal compounds in commercial parenteral lipid emulsions. Nutrients 2012, 4, 904–921. [Google Scholar] [CrossRef]
- Ostlund, R.E., Jr. Phytosterols in human nutrition. Annu. Rev. Nutr. 2002, 22, 533–549. [Google Scholar] [CrossRef]
- Clayton, P.T.; Whitfield, P.; Iyer, K. The role of phytosterols in the pathogenesis of liver complications of pediatric parenteral nutrition. Nutrition 1998, 14, 158–164. [Google Scholar]
- Salen, G.; Ahrens, E.H., Jr.; Grundy, S.M. Metabolism of beta-sitosterol in man. J. Clin. Invest. 1970, 49, 952–967. [Google Scholar] [CrossRef]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar]
- Ostlund, R.E., Jr.; McGill, J.B.; Zeng, C.M.; Covey, D.F.; Stearns, J.; Stenson, W.F.; Spilburg, C.A. Gastrointestinal absorption and plasma kinetics of soy Delta(5)-phytosterols and phytostanols in humans. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E911–E916. [Google Scholar]
- Van Aerde, J.E.; Duerksen, D.R.; Gramlich, L.; Meddings, J.B.; Chan, G.; Thomson, A.B.; Clandinin, M.T. Intravenous fish oil emulsion attenuates total parenteral nutrition-induced cholestasis in newborn piglets. Pediatr. Res. 1999, 45, 202–208. [Google Scholar]
- Iyer, K.R.; Spitz, L.; Clayton, P. BAPS Prize Lecture: New insight into mechanisms of parenteral nutrition-associated cholestasis: Role of plant sterols. British Association of Paediatric Surgeons. J. Pediatr. Surg. 1998, 33, 1–6. [Google Scholar] [CrossRef]
- Clayton, P.T.; Bowron, A.; Mills, K.A.; Massoud, A.; Casteels, M.; Milla, P.J. Phytosterolemia in children with parenteral nutrition-associated cholestatic liver disease. Gastroenterology 1993, 105, 1806–1813. [Google Scholar]
- Ling, W.H.; Jones, P.J. Dietary phytosterols: A review of metabolism, benefits and side effects. Life Sci. 1995, 57, 195–206. [Google Scholar]
- Carter, B.A.; Taylor, O.A.; Prendergast, D.R.; Zimmerman, T.L.; Von Furstenberg, R.; Moore, D.D.; Karpen, S.J. Stigmasterol, a soy lipid-derived phytosterol, is an antagonist of the bile acid nuclear receptor FXR. Pediatr. Res. 2007, 62, 301–306. [Google Scholar] [CrossRef]
- Shefer, S.; Salen, G.; Nguyen, L.; Batta, A.K.; Packin, V.; Tint, G.S.; Hauser, S. Competitive inhibition of bile acid synthesis by endogenous cholestanol and sitosterol in sitosterolemia with xanthomatosis Effect on cholesterol 7 alpha-hydroxylase. J. Clin. Invest. 1988, 82, 1833–1839. [Google Scholar]
- Boberg, K.M.; Akerlund, J.E.; Björkhem, I. Effect of sitosterol on the rate-limiting enzymes in cholesterol synthesis and degradation. Lipids 1989, 24, 9–12. [Google Scholar] [CrossRef]
- Liu, Y.; Binz, J.; Numerick, M.J.; Dennis, S.; Luo, G.; Desai, B.; MacKenzie, K.I.; Mansfield, T.A.; Kliewer, S.A.; Goodwin, B.; Jones, S.A. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J. Clin. Invest. 2003, 112, 1678–1687. [Google Scholar]
- Wanten, G.; Beunk, J.; Naber, A.; Swinkels, D. Tocopherol isoforms in parenteral lipid emulsions and neutrophil activation. Clin. Nutr. 2002, 21, 417–422. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, K.; Hagen, T.M. Oxidants, antioxidants and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar]
- Kohen, R.; Nyska, A. Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Hong, L.; Wang, X.; Wu, J.; Cai, W. Mitochondria-initiated apoptosis triggered by oxidative injury play a role in total parenteral nutrition—associated liver dysfunction in infant rabbit model. J. Pediatr. Surg. 2009, 44, 1712–1718. [Google Scholar] [CrossRef]
- Chung, M.Y.; Yeung, S.F.; Park, H.J.; Volek, J.S.; Bruno, R.S. Dietary α- and γ-tocopherol supplementation attenuates lipopolysaccharide-induced oxidative stress and inflammatory-related responses in an obese mouse model of nonalcoholic steatohepatitis. J. Nutr. Biochem. 2010, 21, 1200–1206. [Google Scholar] [CrossRef]
- Soden, J.S.; Devereaux, M.W.; Haas, J.E.; Gumpricht, E.; Dahl, R.; Gralla, J.; Traber, M.G.; Sokol, R.J. Subcutaneous vitamin E ameliorates liver injury in an in vivo model of steatocholestasis. Hepatology 2007, 46, 485–495. [Google Scholar]
- Becvarova, I.; Saker, K.E.; Swecker, W.S., Jr.; Troy, G.C. Peroxidative protection of parenteral admixture by D-alpha-tocopherol. Vet. Ther. 2005, 6, 280–290. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, M.; Bass, M.; Neushwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Eng. J. Med. 2010, 365, 1675–1685. [Google Scholar]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar]
- Strijbosch, R.A.M.; Lee, S.; Arsenault, D.A.; Andersson, C.; Gura, K.M.; Bistrian, B.R.; Puder, M. Fish oil prevents essential fatty acid deficiency and enhances growth: Clinical and biochemical implications. Metabolism 2008, 57, 698–707. [Google Scholar] [CrossRef]
- De Meijer, V.E.; Le, H.; Meisel, J.A.; Gura, K.M.; Puder, M. Parenteral fish oil as a monotherapy prevents essential fatty acid deficiency in parenteral nutrition-dependent patients. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 212–218. [Google Scholar] [CrossRef]
- Le, H.D.; de Meijer, V.E.; Robinson, E.; Zurakowski, D.; Potemkin, A.K.; Arsenault, D.A.; Fallon, E.M.; Malkan, A.; Bistrian, B.R.; Gura, K.M.; Puder, M. Parenteral fish-oil-based lipid emulsion improves fatty acid profiles and lipids in parenteral nutrition-dependent children. Am. J. Clin. Nutr. 2011, 94, 749–758. [Google Scholar] [CrossRef]
- Diamond, I.R.; Pencharz, P.B.; Feldman, B.M.; Ling, S.C.; Moore, A.M.; Wales, P.W. Novel lipid-based approaches to pediatric intestinal failure-associated liver disease. Arch. Pediatr. Adolesc. Med. 2012, 166, 473–478. [Google Scholar] [CrossRef]
- Nehra, D.; Le, H.D.; Fallon, E.M.; Carlson, S.J.; Woods, D.; White, Y.A.; Pan, A.H.; Guo, L.; Rodig, S.J.; Tilly, J.L.; Rueda, B.R.; Puder, M. Prolonging the female reproductive lifespan and improving egg quality with dietary omega-3 fatty acids. Aging Cell 2012, 11, 1046–1054. [Google Scholar]
- Le, H.D.; Meisel, J.A.; de Meijer, V.E.; Fallon, E.M.; Gura, K.M.; Nose, V.; Bistrian, B.R.; Puder, M. Docosahexaenoic acid and arachidonic acid prevent essential fatty acid deficiency and hepatic steatosis. JPEN J. Parenter. Enteral. Nutr. 2012, 36, 431–441. [Google Scholar]
- Gura, K.M.; Parsons, S.K.; Bechard, L.J.; Henderson, T.; Dorsey, M.; Phipatanakul, W.; Duggan, C.; Puder, M.; Lenders, C. Use of a fish oil-based lipid emulsion to treat essential fatty acid deficiency in a soy allergic patient receiving parenteral nutrition. Clin. Nutr. 2005, 24, 839–847. [Google Scholar] [CrossRef]
- Gura, K.M.; Duggan, C.P.; Collier, S.B.; Jennings, R.W.; Folkman, J.; Bistrian, B.R.; Puder, M. Reversal of parenteral nutrition-associated liver disease in two infants with short bowel syndrome using parenteral fish oil: Implications for future management. Pediatrics 2006, 118, e197–e201. [Google Scholar] [CrossRef]
- Gura, K.M.; Lee, S.; Valim, C.; Zhou, J.; Kim, S.; Modi, B.P.; Arsenault, D.A.; Strijbosch, R.A.; Lopes, S.; Duggan, C.; Puder, M. Safety and efficacy of a fish-oil-based fat emulsion in the treatment of parenteral nutrition-associated liver disease. Pediatrics 2008, 121, 678–686. [Google Scholar]
- Puder, M.; Valim, C.; Meisel, J.A.; Le, H.D.; de Meijer, V.E.; Robinson, E.M.; Zhou, J.; Duggan, C.; Gura, K.M. Parenteral fish oil improves outcomes in patients with parenteral nutrition-associated liver injury. Ann. Surg. 2009, 250, 395–402. [Google Scholar]
- Ekema, G.; Falchetti, D.; Boroni, G.; Tanca, A.R.; Altana, C.; Righetti, L.; Ridella, M.; Gambarotti, M.; Berchich, L. Reversal of severe parenteral nutrition-associated liver disease in an infant with short bowel syndrome using parenteral fish oil (Omega-3 fatty acids). J. Pediatr. Surg. 2008, 43, 1191–1195. [Google Scholar] [CrossRef]
- Cheung, H.M.; Lam, H.S.; Tam, Y.H.; Lee, K.H.; Ng, P.C. Rescue treatment of infants with intestinal failure and parenteral nutrition-associated cholestasis (PNAC) using a parenteral fish-oil-based lipid. Clin. Nutr. 2009, 28, 209–212. [Google Scholar] [CrossRef]
- Strijbosch, R.A.; van den Hoonaard, T.L.; Olieman, J.F.; Escher, J.C.; Alwayn, I.P.; Meijers-Ijsselstijn, H. Fish oil in prolonged parenteral nutrition in children-omega-3-fatty acids have a beneficial effect on the liver. Ned. Tijdschr. Geneeskd. 2010, 154, A2003. [Google Scholar]
- Calhoun, A.W.; Sullivan, J.E. Omegaven for the treatment of parenteral nutrition associated liver disease: A case study. J. Ky. Med. Assoc. 2009, 107, 55–57. [Google Scholar]
- Pironi, L.; Colecchia, A.; Guidetti, M.; Belluzzi, A.; D’Errico, A. Fish oil-based emulsion for the treatment of parenteral nutrition associated liver disease in an adult patient. e-SPEN 2010, 5, e243–e246. [Google Scholar]
- Jurewitsch, B.; Gardiner, G.; Naccarato, M.; Jeejeebhoy, K.N. Omega-3-enriched lipid emulsion for liver salvage in parenteral nutrition induced cholestasis in the adult patient. JPEN J. Parenter. Enteral Nutr. 2011, 35, 386–390. [Google Scholar] [CrossRef]
- Burns, D.L.; Gill, B.M. Reversal of parenteral nutrition-associated liver disease with fish oil-based lipid emulsion (Omegaven) in an adult dependent on home parenteral nutrition. JPEN J. Parenter. Enteral Nutr. 2012, in press. [Google Scholar]
- Mallah, H.S.; Brown, M.R.; Rossi, T.M.; Block, R.C. Parenteral fish oil-associated burr cell anemia. J. Pediatr. 2010, 156, 324–326. [Google Scholar] [CrossRef]
- Grahn, E.P.; Dietz, A.A.; Stefani, S.S.; Donnelly, W.J. Burr cells, hemolytic anemia and cirrhosis. Am. J. Med. 1968, 45, 78–87. [Google Scholar]
- Soden, J.S.; Lovell, M.A.; Brown, K.; Partrick, D.A.; Sokol, R.J. Failure of resolution of portal fibrosis during omega-3 fatty acid lipid emulsion therapy in two patients with irreversible failure. J. Pediatr. 2010, 156, 327–331. [Google Scholar]
- Dimmitt, R.A.; Leadford, A.L.; Bartle, D.; Wilkinson, L.; Manimaran, V.; Harmon, M. Impact of Omega-3 Fatty Acids on Liver Function Test and Liver Histology in Children with Intestinal Failure Associated Intestinal Disease; Pediatric Academic Society: Boston, MA, USA, 2012; E-PAS2012:3830.366. [Google Scholar]
- Diamond, I.R.; Sterescu, A.; Pencharz, P.B.; Kim, J.H.; Wales, P.W. Changing the paradigm: Omegaven for the treatment of liver failure in pediatric short bowel syndrome. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 209–215. [Google Scholar] [CrossRef]
- Muhammed, R.; Bremner, R.; Protheroe, S.; Johnson, T.; Holden, C.; Murphy, S.M. Resolution of parenteral nutrition-associated jaundice on changing from a soybean oil emulsion to a complex mixed-lipid emulsion. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 797–802. [Google Scholar] [CrossRef]
- Tomsits, E.; Pataki, M.; Tölgyesi, A.; Fekete, G.; Rischak, K.; Szollár, L. Safety and efficacy of a lipid emulsion containing a mixture of soybean oil, medium-chain triglycerides, olive oil, and fish oil: A randomised, double-blind clinical trial in premature infants requiring parenteral nutrition. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 514–521. [Google Scholar] [CrossRef]
- Goulet, O.; Antébi, H.; Wolf, C.; Talbotec, C.; Alcindor, L.G.; Corriol, O.; Lamor, M.; Colomb-Jung, V. A new intravenous fat emulsion containing soybean oil, medium-chain triglycerides, olive oil, and fish oil: A single-center, double-blind randomized study on efficacy and safety in pediatric patients receiving home parenteral nutrition. JPEN J. Parenter. Enteral Nutr. 2010, 34, 485–495. [Google Scholar] [CrossRef]
- Colomb, V.; Jobert-Giraud, A.; Lacaille, F.; Goulet, O.; Fournet, J.C.; Ricour, C. Role of lipid emulsions in cholestasis associated with long-term parenteral nutrition in children. JPEN J. Parenter. Enteral. Nutr. 2000, 24, 345–350. [Google Scholar]
- Rubinos, L.H.; Ruth, J.S.; Hawthorne, K.; Abrams, S.A. Reducing Soy-Based Lipid ConcentrationMay Not Prevent the Need for Omega-3 Therapy in Parenteral Nutrition Associated Cholestasis; Pediatric Academic Society: Boston, MA, USA, 2012; E-PAS2012:3830.369. [Google Scholar]
- Nehra, D.; Fallon, E.M.; Carlson, S.J.; Potemkin, A.K.; Hevelone, N.D.; Mitchell, P.D.; Gura, K.M.; Puder, M. Provision of a soy-based intravenous lipid emulsion at 1 g/kg/d does not prevent cholestasis in neonates. JPEN J. Parenter. Enteral Nutr. 2012, in press. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chang, M.I.; Puder, M.; Gura, K.M. The Use of Fish Oil Lipid Emulsion in the Treatment of Intestinal Failure Associated Liver Disease (IFALD). Nutrients 2012, 4, 1828-1850. https://doi.org/10.3390/nu4121828
Chang MI, Puder M, Gura KM. The Use of Fish Oil Lipid Emulsion in the Treatment of Intestinal Failure Associated Liver Disease (IFALD). Nutrients. 2012; 4(12):1828-1850. https://doi.org/10.3390/nu4121828
Chicago/Turabian StyleChang, Melissa I., Mark Puder, and Kathleen M. Gura. 2012. "The Use of Fish Oil Lipid Emulsion in the Treatment of Intestinal Failure Associated Liver Disease (IFALD)" Nutrients 4, no. 12: 1828-1850. https://doi.org/10.3390/nu4121828
APA StyleChang, M. I., Puder, M., & Gura, K. M. (2012). The Use of Fish Oil Lipid Emulsion in the Treatment of Intestinal Failure Associated Liver Disease (IFALD). Nutrients, 4(12), 1828-1850. https://doi.org/10.3390/nu4121828