Induction of Cancer Cell Death by Isoflavone: The Role of Multiple Signaling Pathways

Abstract
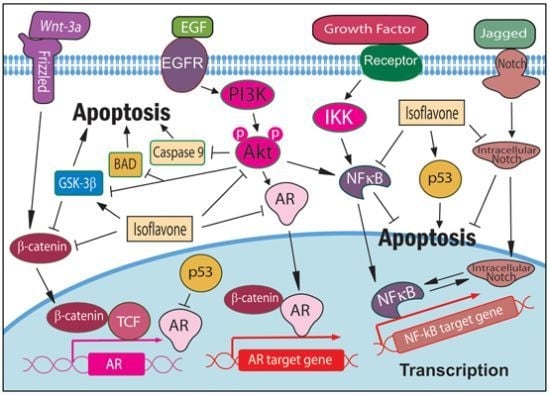
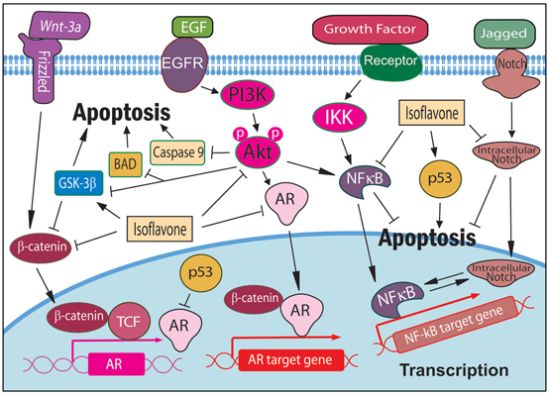
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Deregulation of Cellular Signaling and Apoptosis Pathway in Cancer Cells

3. Regulation of Cellular Signaling to Induce Apoptosis by Isoflavone
3.1. Isoflavone Induces Apoptosis through the Regulation of NF-κB Signaling
3.2. Isoflavone Induces Apoptosis through the Regulation of Akt Signaling
3.3. Isoflavone Induces Apoptosis through the Regulation of MAPK Signaling
3.4. Isoflavone Induces Apoptosis through the Regulation of Wnt Signaling
3.5. Isoflavone Induces Apoptosis through the Regulation of Notch Signaling
3.6. Isoflavone Induces Apoptosis through the Regulation of p53 Signaling
3.7. Isoflavone Induces Apoptosis through the Regulation of AR Signaling
3.8. Isoflavone Regulates miRNAs, Leading to Increased Drug Sensitivity and Cancer Cell Death
4. Summary and Perspectives
Acknowledgements
References
- Meier, P.; Finch, A.; Evan, G. Apoptosis in development. Nature 2000, 407, 796–801. [Google Scholar]
- Zhou, Z.; Sun, X.; Kang, Y.J. Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am. J. Pathol. 2001, 159, 329–338. [Google Scholar]
- Shi, Y. Caspase activation: Revisiting the induced proximity model. Cell 2004, 117, 855–858. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [PubMed]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Iyer, S.; Boulares, H.; Smulson, M.E. Involvement of PARP and poly(ADP-ribosyl)ation in the early stages of apoptosis and DNA replication. Mol. Cell. Biochem. 1999, 193, 137–148. [Google Scholar]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar]
- Kitada, S.; Pedersen, I.M.; Schimmer, A.D.; Reed, J.C. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene 2002, 21, 3459–3474. [Google Scholar]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar]
- Yakovlev, A.G.; Wang, G.; Stoica, B.A.; Boulares, H.A.; Spoonde, A.Y.; Yoshihara, K.; Smulson, M.E. A role of the Ca2+/Mg2+-dependent endonuclease in apoptosis and its inhibition by Poly(ADP-ribose) polymerase. J. Biol. Chem. 2000, 275, 21302–21308. [Google Scholar]
- Guo, Y.; Srinivasula, S.M.; Druilhe, A.; Fernandes-Alnemri, T.; Alnemri, E.S. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J. Biol. Chem. 2002, 277, 13430–13437. [Google Scholar]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar]
- Kasibhatla, S.; Tseng, B. Why target apoptosis in cancer treatment? Mol. Cancer Ther. 2003, 2, 573–580. [Google Scholar] [PubMed]
- Shaw, A.S.; Filbert, E.L. Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol. 2009, 9, 47–56. [Google Scholar]
- Martin, G.S. Cell signaling and cancer. Cancer Cell 2003, 4, 167–174. [Google Scholar]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar]
- Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 2007, 7, 165–168. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar]
- Lee, M.M.; Gomez, S.L.; Chang, J.S.; Wey, M.; Wang, R.T.; Hsing, A.W. Soy and isoflavone consumption in relation to prostate cancer risk in China. Cancer Epidemiol. Biomark. Prev. 2003, 12, 665–668. [Google Scholar]
- Mukhtar, H.; Ahmad, N. Green tea in chemoprevention of cancer. Toxicol. Sci. 1999, 52, 111–117. [Google Scholar]
- Smith-Warner, S.A.; Spiegelman, D.; Yaun, S.S.; Albanes, D.; Beeson, W.L.; van den Brandt, P.A.; Feskanich, D.; Folsom, A.R.; Fraser, G.E.; Freudenheim, J.L.; et al. Fruits, vegetables and lung cancer: a pooled analysis of cohort studies. Int. J. Cancer 2003, 107, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar]
- Khan, N.; Afaq, F.; Mukhtar, H. Apoptosis by dietary factors: The suicide solution for delaying cancer growth. Carcinogenesis 2007, 28, 233–239. [Google Scholar]
- Lamartiniere, C.A.; Cotroneo, M.S.; Fritz, W.A.; Wang, J.; Mentor-Marcel, R.; Elgavish, A. Genistein chemoprevention: timing and mechanisms of action in murine mammary and prostate. J. Nutr. 2002, 132, 552S–558S. [Google Scholar]
- Li, Y.; Li, X.; Sarkar, F.H. Gene expression profiles of I3C- and DIM-treated PC3 human prostate cancer cells determined by cDNA microarray analysis. J. Nutr. 2003, 133, 1011–1019. [Google Scholar]
- Gupta, S.; Hussain, T.; Mukhtar, H. Molecular pathway for (−)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch. Biochem. Biophys. 2003, 410, 177–185. [Google Scholar]
- Hastak, K.; Gupta, S.; Ahmad, N.; Agarwal, M.K.; Agarwal, M.L.; Mukhtar, H. Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene 2003, 22, 4851–4859. [Google Scholar]
- Dixon, R.A.; Ferreira, D. Genistein. Phytochemistry 2002, 60, 205–211. [Google Scholar]
- Li, Y.; Sarkar, F.H. Down-regulation of invasion and angiogenesis-related genes identified by cDNA microarray analysis of PC3 prostate cancer cells treated with genistein. Cancer Lett. 2002, 186, 157–164. [Google Scholar]
- Storz, P.; Toker, A. NF-kappaB signaling-an alternate pathway for oxidative stress responses. Cell Cycle 2003, 2, 9–10. [Google Scholar]
- Lin, A.; Karin, M. NF-kappaB in cancer: a marked target. Semin. Cancer Biol. 2003, 13, 107–114. [Google Scholar]
- Haefner, B. NF-kappaB: Arresting a major culprit in cancer. Drug Discov. Today 2002, 7, 653–663. [Google Scholar]
- Orlowski, R.Z.; Baldwin, A.S. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002, 8, 385–389. [Google Scholar]
- Clarkson, R.W.; Heeley, J.L.; Chapman, R.; Aillet, F.; Hay, R.T.; Wyllie, A.; Watson, C.J. NF-kappaB inhibits apoptosis in murine mammary epithelia. J. Biol. Chem. 2000, 275, 12737–12742. [Google Scholar]
- Arlt, A.; Vorndamm, J.; Breitenbroich, M.; Folsch, U.R.; Kalthoff, H.; Schmidt, W.E.; Schafer, H. Inhibition of NF-kappaB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene 2001, 20, 859–868. [Google Scholar]
- Mitsiades, C.S.; Mitsiades, N.; Poulaki, V.; Schlossman, R.; Akiyama, M.; Chauhan, D.; Hideshima, T.; Treon, S.P.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: Therapeutic implications. Oncogene 2002, 21, 5673–5683. [Google Scholar]
- Rogers, R.; Ouellet, G.; Brown, C.; Moyer, B.; Rasoulpour, T.; Hixon, M. Cross-talk between the Akt and NF-kappaB signaling pathways inhibits MEHP-induced germ cell apoptosis. Toxicol. Sci. 2008, 106, 497–508. [Google Scholar]
- Clarke, R.B. p27KIP1 phosphorylation by PKB/Akt leads to poor breast cancer prognosis. Breast Cancer Res. 2003, 5, 162–163. [Google Scholar]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherap. Leukemia 2003, 17, 590–603. [Google Scholar]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar]
- Rommel, C.; Clarke, B.A.; Zimmermann, S.; Nunez, L.; Rossman, R.; Reid, K.; Moelling, K.; Yancopoulos, G.D.; Glass, D.J. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 1999, 286, 1738–1741. [Google Scholar]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar]
- Hill, M.M.; Hemmings, B.A. Inhibition of protein kinase B/Akt. implications for cancer therapy. Pharmacol. Ther. 2002, 93, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Sebolt-Leopold, J.S. Development of anticancer drugs targeting the MAP kinase pathway. Oncogene 2000, 19, 6594–6599. [Google Scholar]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar]
- Lunghi, P.; Giuliani, N.; Mazzera, L.; Lombardi, G.; Ricca, M.; Corradi, A.; Cantoni, A.M.; Salvatore, L.; Riccioni, R.; Costanzo, A.; et al. Targeting MEK/MAPK signal transduction module potentiates ATO-induced apoptosis in multiple myeloma cells through multiple signaling pathways. Blood 2008, 112, 2450–2462. [Google Scholar] [PubMed]
- Zhang, Y.; Ting, A.T.; Marcu, K.B.; Bliska, J.B. Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J. Immunol. 2005, 174, 7939–7949. [Google Scholar]
- van Laethem, A.; van Kelst, S.; Lippens, S.; Declercq, W.; Vandenabeele, P.; Janssens, S.; Vandenheede, J.R.; Garmyn, M.; Agostinis, P. Activation of p38 MAPK is required for Bax translocation to mitochondria, cytochrome c release and apoptosis induced by UVB irradiation in human keratinocytes. FASEB J. 2004, 18, 1946–1948. [Google Scholar]
- Tanaka, Y.; Gavrielides, M.V.; Mitsuuchi, Y.; Fujii, T.; Kazanietz, M.G. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J. Biol. Chem. 2003, 278, 33753–33762. [Google Scholar]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar]
- Behrens, J. Control of beta-catenin signaling in tumor development. Ann. N. Y. Acad. Sci. 2000, 910, 21–33. [Google Scholar]
- Peifer, M.; Polakis, P. Wnt signaling in oncogenesis and embryogenesis-a look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar]
- Dehner, M.; Hadjihannas, M.; Weiske, J.; Huber, O.; Behrens, J. Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J. Biol. Chem. 2008, 283, 19201–19210. [Google Scholar]
- Almeida, M.; Han, L.; Bellido, T.; Manolagas, S.C.; Kousteni, S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J. Biol. Chem. 2005, 280, 41342–41351. [Google Scholar]
- He, B.; Reguart, N.; You, L.; Mazieres, J.; Xu, Z.; Lee, A.Y.; Mikami, I.; McCormick, F.; Jablons, D.M. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 2005, 24, 3054–3058. [Google Scholar]
- You, L.; He, B.; Xu, Z.; Uematsu, K.; Mazieres, J.; Fujii, N.; Mikami, I.; Reguart, N.; McIntosh, J.K.; Kashani-Sabet, M.; McCormick, F.; Jablons, D.M. An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant melanoma cells and inhibits tumor growth. Cancer Res. 2004, 64, 5385–5389. [Google Scholar]
- Dihlmann, S.; von Knebel, D.M. Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int. J. Cancer 2005, 113, 515–524. [Google Scholar]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar]
- Zardawi, S.J.; O’Toole, S.A.; Sutherland, R.L.; Musgrove, E.A. Dysregulation of hedgehog, Wnt and notch signalling pathways in breast cancer. Histol. Histopathol. 2009, 24, 385–398. [Google Scholar]
- Wang, Z.; Li, Y.; Banerjee, S.; Sarkar, F.H. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009, 279, 8–12. [Google Scholar]
- Wang, Z.; Banerjee, S.; Li, Y.; Rahman, K.M.; Zhang, Y.; Sarkar, F.H. Down-regulation of notch-1 inhibits invasion by inactivation of nuclear factor-kappaB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cell. Cancer Res. 2006, 66, 2778–2784. [Google Scholar]
- Meurette, O.; Stylianou, S.; Rock, R.; Collu, G.M.; Gilmore, A.P.; Brennan, K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009, 69, 5015–5022. [Google Scholar]
- Liu, W.H.; Hsiao, H.W.; Tsou, W.I.; Lai, M.Z. Notch inhibits apoptosis by direct interference with XIAP ubiquitination and degradation. EMBO J. 2007, 26, 1660–1669. [Google Scholar]
- Wang, Z.; Zhang, Y.; Li, Y.; Banerjee, S.; Liao, J.; Sarkar, F.H. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol. Cancer Ther. 2006, 5, 483–493. [Google Scholar]
- Wang, Z.; Li, Y.; Banerjee, S.; Kong, D.; Ahmad, A.; Nogueira, V.; Hay, N.; Sarkar, F.H. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J. Cell. Biochem. 2010, 109, 726–736. [Google Scholar]
- Nefedova, Y.; Sullivan, D.M.; Bolick, S.C.; Dalton, W.S.; Gabrilovich, D.I. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood 2008, 111, 2220–2229. [Google Scholar]
- Zweidler-McKay, P.A.; He, Y.; Xu, L.; Rodriguez, C.G.; Karnell, F.G.; Carpenter, A.C.; Aster, J.C.; Allman, D.; Pear, W.S. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood 2005, 106, 3898–3906. [Google Scholar]
- Chadwick, N.; Fennessy, C.; Nostro, M.C.; Baron, M.; Brady, G.; Buckle, A.M. Notch induces cell cycle arrest and apoptosis in human erythroleukaemic TF-1 cells. Blood Cells Mol. Dis. 2008, 41, 270–277. [Google Scholar]
- Manic, S.; Gatti, L.; Carenini, N.; Fumagalli, G.; Zunino, F.; Perego, P. Mechanisms controlling sensitivity to platinum complexes: Role of p53 and DNA mismatch repair. Curr. Cancer Drug Targets 2003, 3, 21–29. [Google Scholar]
- Luke, M.C.; Coffey, D.S. Human androgen receptor binding to the androgen response element of prostate specific antigen. J. Androl. 1994, 15, 41–51. [Google Scholar]
- Montgomery, J.S.; Price, D.K.; Figg, W.D. The androgen receptor gene and its influence on the development and progression of prostate cancer. J. Pathol. 2001, 195, 138–146. [Google Scholar]
- Kupelian, P.; Katcher, J.; Levin, H.; Zippe, C.; Klein, E. Correlation of clinical and pathologic factors with rising prostate-specific antigen profiles after radical prostatectomy alone for clinically localized prostate cancer. Urology 1996, 48, 249–260. [Google Scholar]
- Sato, N.; Gleave, M.E.; Bruchovsky, N.; Rennie, P.S.; Goldenberg, S.L.; Lange, P.H.; Sullivan, L.D. Intermittent androgen suppression delays progression to androgen-independent regulation of prostate-specific antigen gene in the LNCaP prostate tumour model. J. Steroid Biochem. Mol. Biol. 1996, 58, 139–146. [Google Scholar]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar]
- Lin, Y.; Kokontis, J.; Tang, F.; Godfrey, B.; Liao, S.; Lin, A.; Chen, Y.; Xiang, J. Androgen and its receptor promote Bax-mediated apoptosis. Mol. Cell. Biol. 2006, 26, 1908–1916. [Google Scholar]
- Chiu, F.L.; Lin, J.K. Downregulation of androgen receptor expression by luteolin causes inhibition of cell proliferation and induction of apoptosis in human prostate cancer cells and xenografts. Prostate 2008, 68, 61–71. [Google Scholar]
- Chen, D.; Cui, Q.C.; Yang, H.; Barrea, R.A.; Sarkar, F.H.; Sheng, S.; Yan, B.; Reddy, G.P.; Dou, Q.P. Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007, 67, 1636–1644. [Google Scholar] [PubMed]
- Hebert, J.R.; Hurley, T.G.; Olendzki, B.C.; Teas, J.; Ma, Y.; Hampl, J.S. Nutritional and socioeconomic factors in relation to prostate cancer mortality: a cross-national study. J. Natl. Cancer Inst. 1998, 90, 1637–1647. [Google Scholar]
- Jacobsen, B.K.; Knutsen, S.F.; Fraser, G.E. Does high soy milk intake reduce prostate cancer incidence? The adventist health study (United States). Cancer Causes Control 1998, 9, 553–557. [Google Scholar]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Wojewoda, C.; Miele, L.; Sarkar, F.H. Down-regulation of Notch-1 is associated with Akt and FoxM1 in inducing cell growth inhibition and apoptosis in prostate cancer cells. J. Cell. Biochem. 2010, 112, 78–88. [Google Scholar]
- Alhasan, S.A.; Aranha, O.; Sarkar, F.H. Genistein elicits pleiotropic molecular effects on head and neck cancer cells. Clin. Cancer Res. 2001, 7, 4174–4181. [Google Scholar]
- Li, Y.; Upadhyay, S.; Bhuiyan, M.; Sarkar, F.H. Induction of apoptosis in breast cancer cells MDA-MB-231 by genistein. Oncogene 1999, 18, 3166–3172. [Google Scholar]
- Li, Y.; Sarkar, F.H. Inhibition of nuclear factor kappaB activation in PC3 cells by genistein is mediated via Akt signaling pathway. Clin. Cancer Res. 2002, 8, 2369–2377. [Google Scholar]
- Li, Y.; Ellis, K.L.; Ali, S.; El-Rayes, B.F.; Nedeljkovic-Kurepa, A.; Kucuk, O.; Philip, P.A.; Sarkar, F.H. Apoptosis-inducing effect of chemotherapeutic agents is potentiated by soy isoflavone genistein, a natural inhibitor of NF-kappaB in BxPC-3 pancreatic cancer cell line. Pancreas 2004, 28, e90–e95. [Google Scholar]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar]
- Li, Y.; Wang, Z.; Kong, D.; Li, R.; Sarkar, S.H.; Sarkar, F.H. Regulation of Akt/FOXO3a/GSK-3beta/AR signaling network by isoflavone in prostate cancer cells. J. Biol. Chem. 2008, 283, 27707–27716. [Google Scholar]
- Lian, F.; Bhuiyan, M.; Li, Y.W.; Wall, N.; Kraut, M.; Sarkar, F.H. Genistein-induced G2-M arrest, p21WAF1 upregulation, and apoptosis in a non-small-cell lung cancer cell line. Nutr. Cancer 1998, 31, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Baxa, D.M.; Yoshimura, F.K. Genistein reduces NF-kappa B in T lymphoma cells via a caspase-mediated cleavage of I kappa B alpha. Biochem. Pharmacol. 2003, 66, 1009–1018. [Google Scholar]
- Gong, L.; Li, Y.; Nedeljkovic-Kurepa, A.; Sarkar, F.H. Inactivation of NF-kappaB by genistein is mediated via Akt signaling pathway in breast cancer cells. Oncogene 2003, 22, 4702–4709. [Google Scholar]
- Lee, Y.K.; Park, O.J. Soybean isoflavone genistein regulates apoptosis through NF-kappaB dependent and independent pathways. Exp. Toxicol. Pathol. 2011. [Google Scholar] [CrossRef]
- Kole, L.; Giri, B.; Manna, S.K.; Pal, B.; Ghosh, S. Biochanin-A, an isoflavon, showed anti-proliferative and anti-inflammatory activities through the inhibition of iNOS expression, p38-MAPK and ATF-2 phosphorylation and blocking NFkappaB nuclear translocation. Eur. J. Pharmacol. 2011, 653, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.N.; Kucuk, O.; Djuric, Z.; Sarkar, F.H. Soy isoflavone supplementation in healthy men prevents NF-kappa B activation by TNF-alpha in blood lymphocytes. Free Radic. Biol. Med. 2001, 30, 1293–1302. [Google Scholar]
- Satoh, H.; Nishikawa, K.; Suzuki, K.; Asano, R.; Virgona, N.; Ichikawa, T.; Hagiwara, K.; Yano, T. Genistein, a soy isoflavone, enhances necrotic-like cell death in a breast cancer cell treated with a chemotherapeutic agent. Res. Commun. Mol. Pathol. Pharmacol. 2003, 113-114, 149-158. [Google Scholar]
- Khoshyomn, S.; Manske, G.C.; Lew, S.M.; Wald, S.L.; Penar, P.L. Synergistic action of genistein and cisplatin on growth inhibition and cytotoxicity of human medulloblastoma cells. Pediatr. Neurosurg. 2000, 33, 123–131. [Google Scholar]
- Singh-Gupta, V.; Zhang, H.; Yunker, C.K.; Ahmad, Z.; Zwier, D.; Sarkar, F.H.; Hillman, G.G. Daidzein effect on hormone refractory prostate cancer in vitro and in vivo compared to genistein and soy extract: potentiation of radiotherapy. Pharm. Res. 2010, 27, 1115–1127. [Google Scholar]
- Singh-Gupta, V.; Zhang, H.; Banerjee, S.; Kong, D.; Raffoul, J.J.; Sarkar, F.H.; Hillman, G.G. Radiation-induced HIF-1alpha cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int. J. Cancer 2009, 124, 1675–1684. [Google Scholar]
- Ahmad, I.U.; Forman, J.D.; Sarkar, F.H.; Hillman, G.G.; Heath, E.; Vaishampayan, U.; Cher, M.L.; Andic, F.; Rossi, P.J.; Kucuk, O. Soy isoflavones in conjunction with radiation therapy in patients with prostate cancer. Nutr. Cancer 2010, 62, 996–1000. [Google Scholar]
- Ma, Y.; Wang, J.; Liu, L.; Zhu, H.; Chen, X.; Pan, S.; Sun, X.; Jiang, H. Genistein potentiates the effect of arsenic trioxide against human hepatocellular carcinoma: Role of Akt and nuclear factor-kappaB. Cancer Lett. 2011, 301, 75–84. [Google Scholar]
- Privat, M.; Aubel, C.; Arnould, S.; Communal, Y.; Ferrara, M.; Bignon, Y.J. AKT and p21 WAF1/CIP1 as potential genistein targets in BRCA1-mutant human breast cancer cell lines. Anticancer Res. 2010, 30, 2049–2054. [Google Scholar]
- Huang, X.; Chen, S.; Xu, L.; Liu, Y.; Deb, D.K.; Platanias, L.C.; Bergan, R.C. Genistein inhibits p38 map kinase activation, matrix metalloproteinase type 2, and cell invasion in human prostate epithelial cell. Cancer Res. 2005, 65, 3470–3478. [Google Scholar]
- Jin, C.Y.; Park, C.; Kim, G.Y.; Lee, S.J.; Kim, W.J.; Choi, Y.H. Genistein enhances TRAIL-induced apoptosis through inhibition of p38 MAPK signaling in human hepatocellular carcinoma Hep3B cells. Chem. Biol. Interact. 2009, 180, 143–150. [Google Scholar]
- Shen, J.; Tai, Y.C.; Zhou, J.; Stephen Wong, C.H.; Cheang, P.T.; Fred Wong, W.S.; Xie, Z.; Khan, M.; Han, J.H.; Chen, C.S. Synergistic antileukemia effect of genistein and chemotherapy in mouse xenograft model and potential mechanism through MAPK signaling. Exp. Hematol. 2007, 35, 75–83. [Google Scholar]
- Sánchez, Y.; Calle, C.; de Blas, E.; Aller, P. Modulation of arsenic trioxide-induced apoptosis by genistein and functionally related agents in U937 human leukaemia cells. Regulation by ROS and mitogen-activated protein kinases. Chem. Biol. Interact. 2009, 182, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, Y.; Amran, D.; Fernandez, C.; de Blas, E.; Aller, P. Genistein selectively potentiates arsenic trioxide-induced apoptosis in human leukemia cells via reactive oxygen species generation and activation of reactive oxygen species-inducible protein kinases (p38-MAPK, AMPK). Int. J. Cancer 2008, 123, 1205–1214. [Google Scholar]
- Su, Y.; Simmen, R.C. Soy isoflavone genistein up-regulates epithelial adhesion molecule E-cadherin expression and attenuates beta-catenin signaling in mammary epithelial cells. Carcinogenesis 2009, 30, 331–339. [Google Scholar]
- Su, Y.; Simmen, F.A.; Xiao, R.; Simmen, R.C. Expression profiling of rat mammary epithelial cells reveals candidate signaling pathways in dietary protection from mammary tumors. Physiol. Genomics 2007, 30, 8–16. [Google Scholar]
- Wagner, J.; Lehmann, L. Estrogens modulate the gene expression of Wnt-7a in cultured endometrial adenocarcinoma cells. Mol. Nutr. Food Res. 2006, 50, 368–372. [Google Scholar]
- Janardhanan, R.; Banik, N.L.; Ray, S.K. N-Myc down regulation induced differentiation, early cell cycle exit, and apoptosis in human malignant neuroblastoma cells having wild type or mutant p5. Biochem. Pharmacol. 2009, 78, 1105–1114. [Google Scholar]
- Lian, F.; Li, Y.; Bhuiyan, M.; Sarkar, F.H. p53-independent apoptosis induced by genistein in lung cancer cells. Nutr. Cancer 1999, 33, 125–131. [Google Scholar]
- Constantinou, A.I.; Kamath, N.; Murley, J.S. Genistein inactivates Bcl-2, delays the G2/M phase of the cell cycle, and induces apoptosis of human breast adenocarcinoma MCF-7 cell. Eur. J. Cancer 1998, 34, 1927–1934. [Google Scholar]
- Tokalov, S.V.; Abramyuk, A.M.; Abolmaali, N.D. Protection of p53 wild type cells from taxol by genistein in the combined treatment of lung cancer. Nutr. Cancer 2010, 62, 795–801. [Google Scholar]
- Roy, C.S.; Karmakar, S.; Banik, N.L.; Ray, S.K. Synergistic efficacy of sorafenib and genistein in growth inhibition by down regulating angiogenic and survival factors and increasing apoptosis through upregulation of p53 and p21 in malignant neuroblastoma cells having N-Myc amplification or non-amplification. Invest. New Drugs 2010, 28, 812–824. [Google Scholar]
- Vinall, R.L.; Hwa, K.; Ghosh, P.; Pan, C.X.; Lara, P.N., Jr.; de Vere White, R.W. Combination treatment of prostate cancer cell lines with bioactive soy isoflavones and perifosine causes increased growth arrest and/or apoptosis. Clin. Cancer Res. 2007, 13, 6204–6216. [Google Scholar]
- Davis, J.N.; Kucuk, O.; Sarkar, F.H. Expression of prostate-specific antigen is transcriptionally regulated by genistein in prostate cancer cells. Mol. Carcinog. 2002, 34, 91–101. [Google Scholar]
- Tepper, C.G.; Vinall, R.L.; Wee, C.B.; Xue, L.; Shi, X.B.; Burich, R.; Mack, P.C.; de Vere White, R.W. GCP-mediated growth inhibition and apoptosis of prostate cancer cells via androgen receptor-dependent and -independent mechanisms. Prostate 2007, 67, 521–535. [Google Scholar]
- Fritz, W.A.; Wang, J.; Eltoum, I.E.; Lamartiniere, C.A. Dietary genistein down-regulates androgen and estrogen receptor expression in the rat prostate. Mol. Cell. Endocrinol. 2002, 186, 89–99. [Google Scholar]
- Li, Y.; VandenBoom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar]
- Tsang, W.P.; Kwok, T.T. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J. Nutr. Biochem. 2010, 21, 140–146. [Google Scholar]
- Chen, Y.; Zaman, M.S.; Deng, G.; Majid, S.; Saini, S.; Liu, J.; Tanaka, Y.; Dahiya, R. MicroRNAs 221/222 and genistein-mediated regulation of ARHI tumor suppressor gene in prostate cancer. Cancer Prev. Res. (Phila.) 2011, 4, 76–86. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrial.org. Available online: http://www.clinicaltrials.gov/ct2/results?term=Isoflavone+AND+cancer (accessed on 26 September 2011).
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Y.; Kong, D.; Bao, B.; Ahmad, A.; Sarkar, F.H. Induction of Cancer Cell Death by Isoflavone: The Role of Multiple Signaling Pathways. Nutrients 2011, 3, 877-896. https://doi.org/10.3390/nu3100877
Li Y, Kong D, Bao B, Ahmad A, Sarkar FH. Induction of Cancer Cell Death by Isoflavone: The Role of Multiple Signaling Pathways. Nutrients. 2011; 3(10):877-896. https://doi.org/10.3390/nu3100877
Chicago/Turabian StyleLi, Yiwei, Dejuan Kong, Bin Bao, Aamir Ahmad, and Fazlul H. Sarkar. 2011. "Induction of Cancer Cell Death by Isoflavone: The Role of Multiple Signaling Pathways" Nutrients 3, no. 10: 877-896. https://doi.org/10.3390/nu3100877
APA StyleLi, Y., Kong, D., Bao, B., Ahmad, A., & Sarkar, F. H. (2011). Induction of Cancer Cell Death by Isoflavone: The Role of Multiple Signaling Pathways. Nutrients, 3(10), 877-896. https://doi.org/10.3390/nu3100877