
Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Procedure
2.1. Animals
2.2. Apparatus
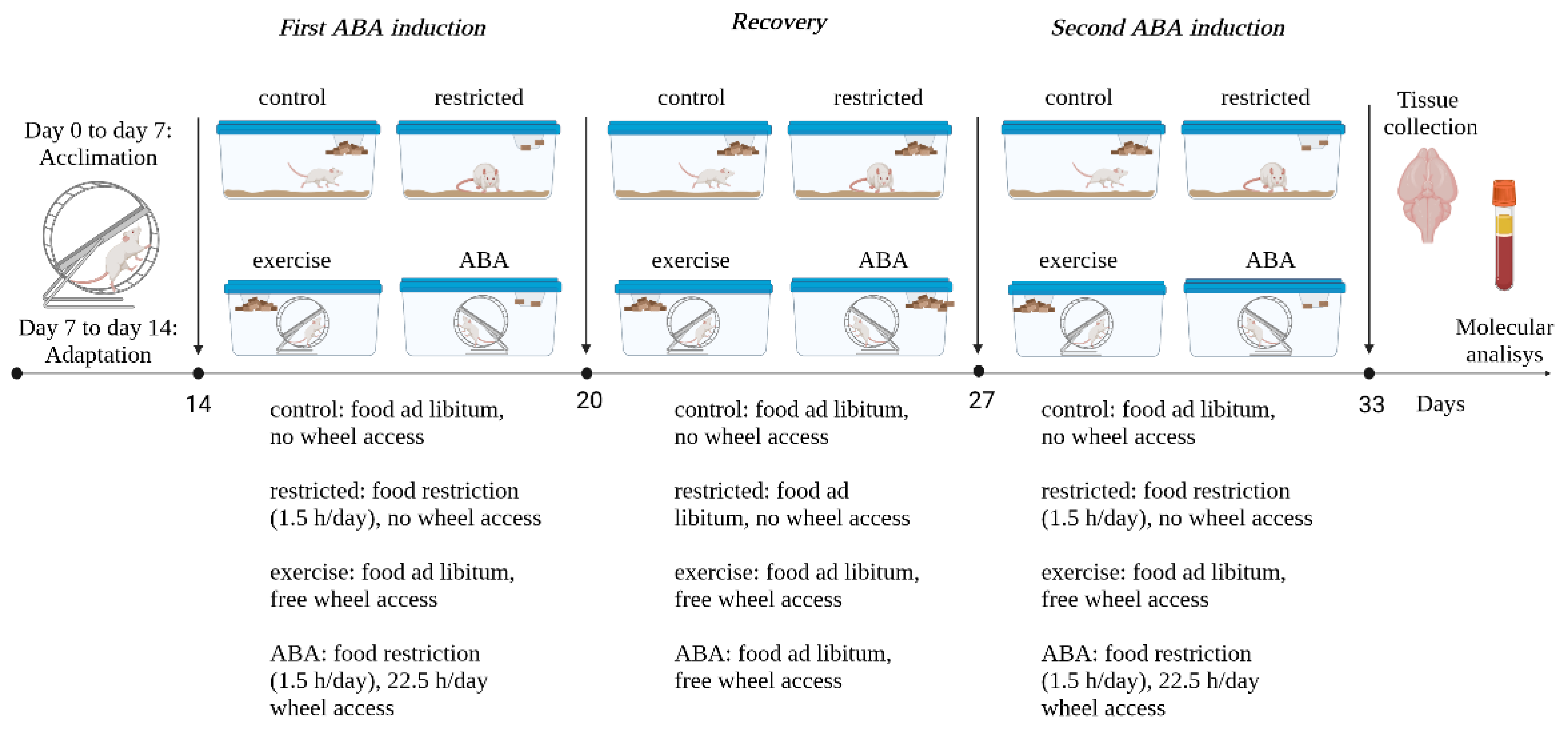
2.3. Experimental Design
2.4. Plasma Adiponectin Level Assay—ELISA
2.5. Tissue Lysate and Western Blotting
2.6. Immunoprecipitation
2.7. Statistical Analysis
3. Results
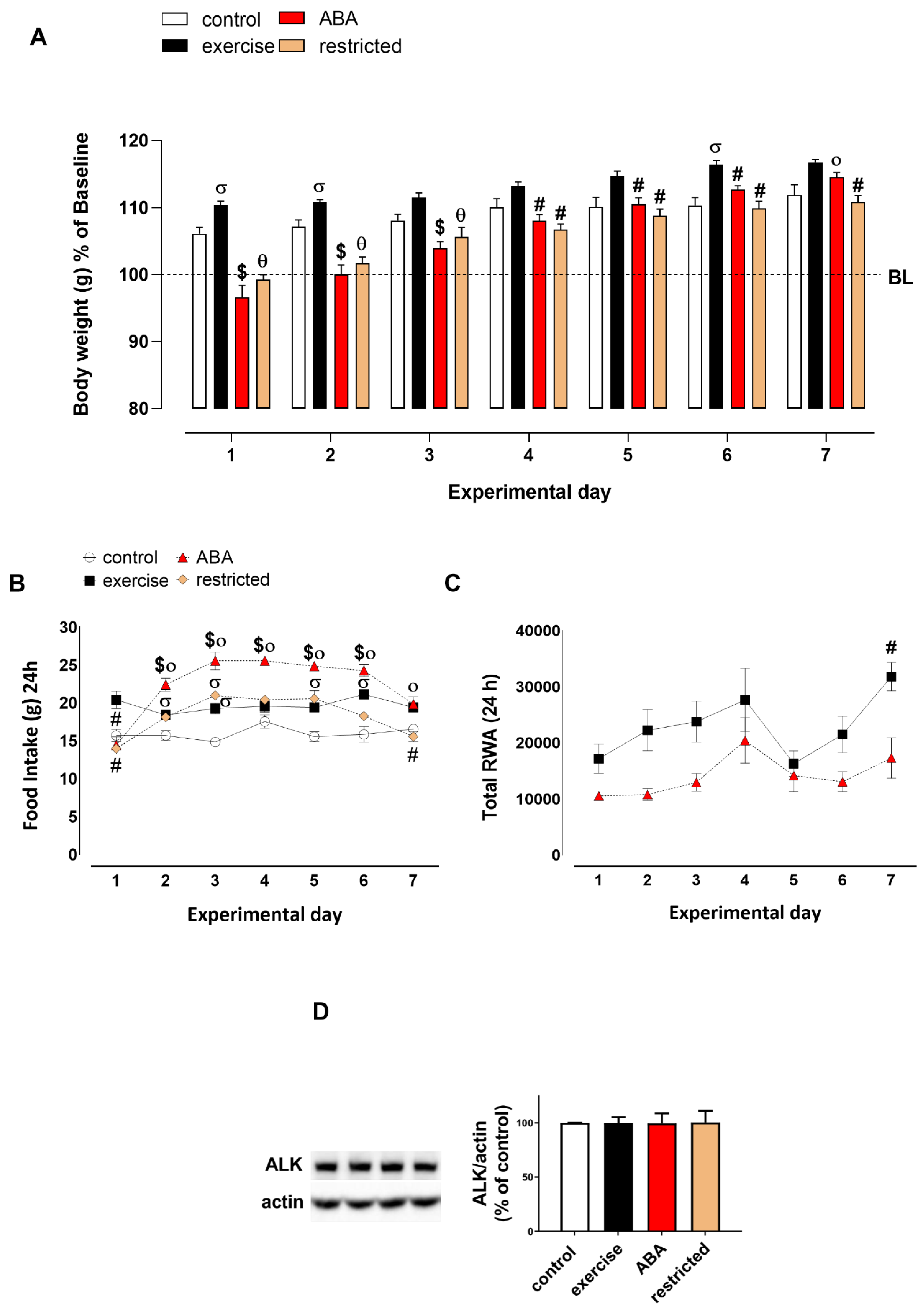
3.1. First ABA Induction
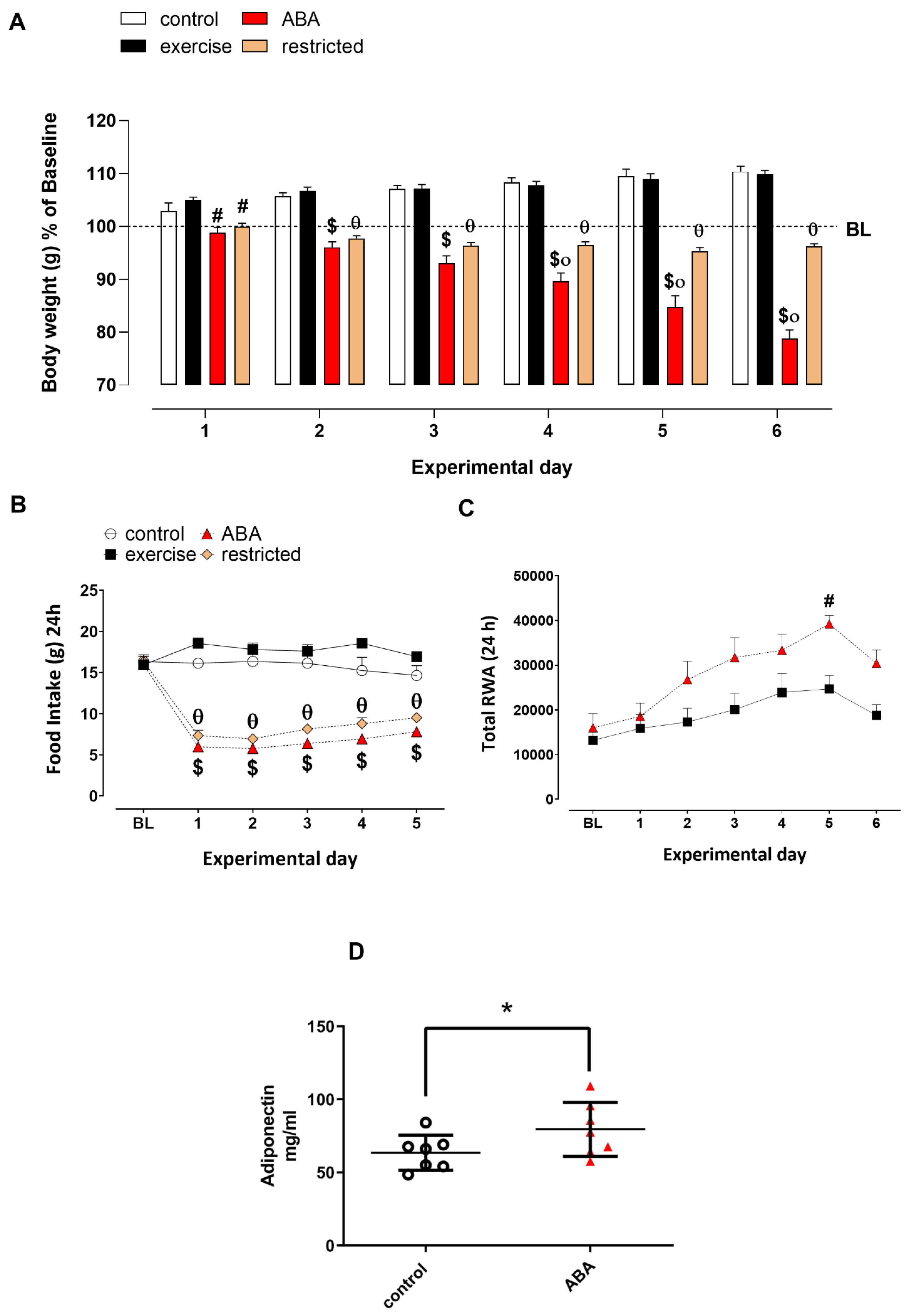
3.1.1. Body Weight, Food Intake, and RWA
3.1.2. Adiponectin Levels
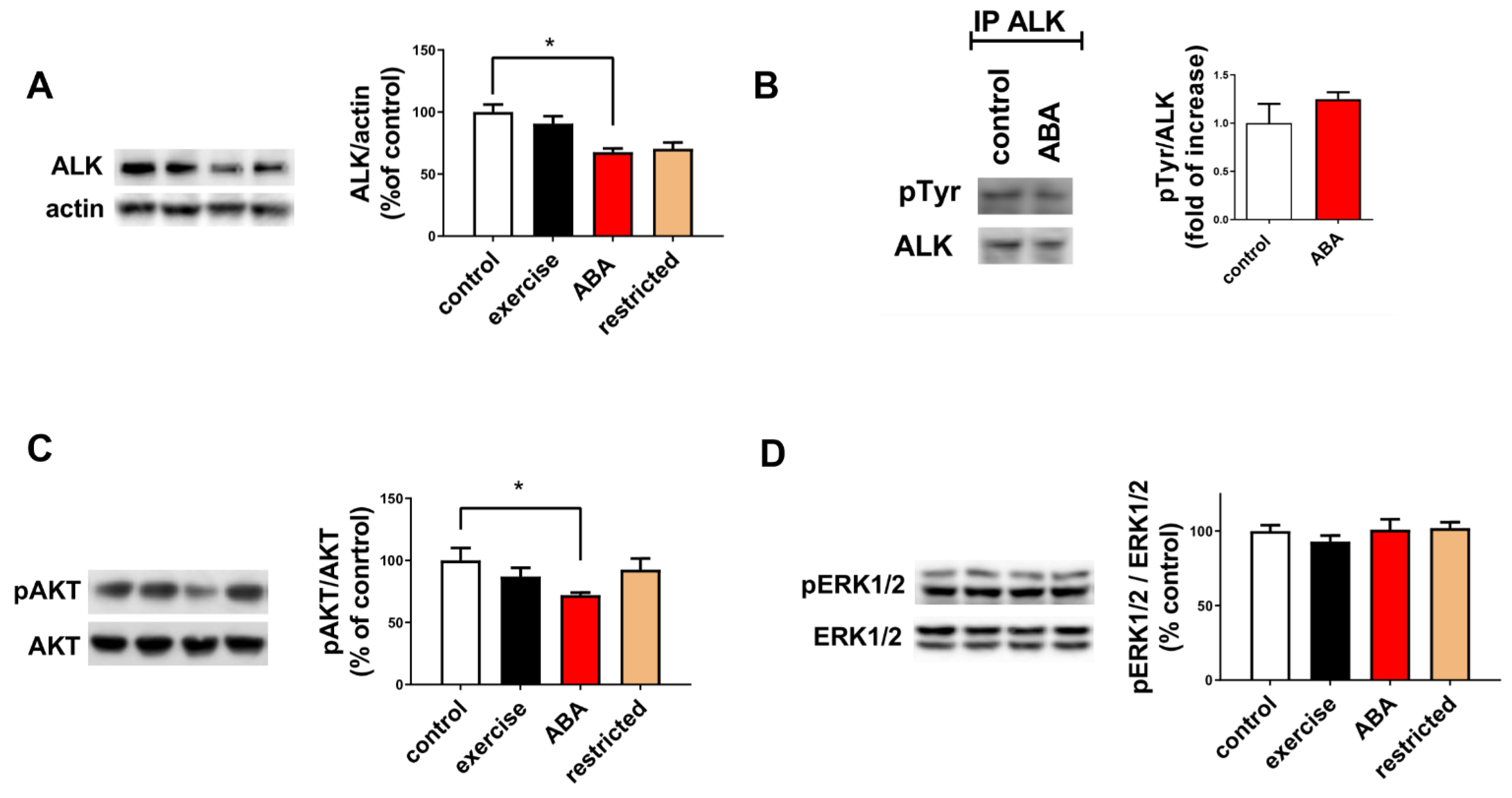
3.1.3. ALK Receptor Signaling
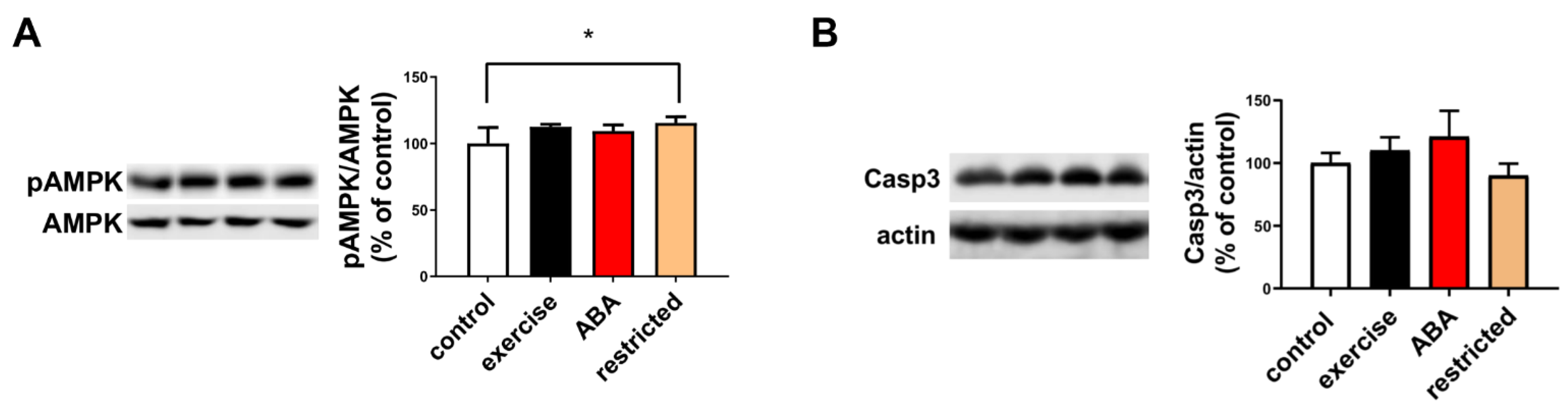
3.1.4. AMPK Phosphorylation and Caspase Activation
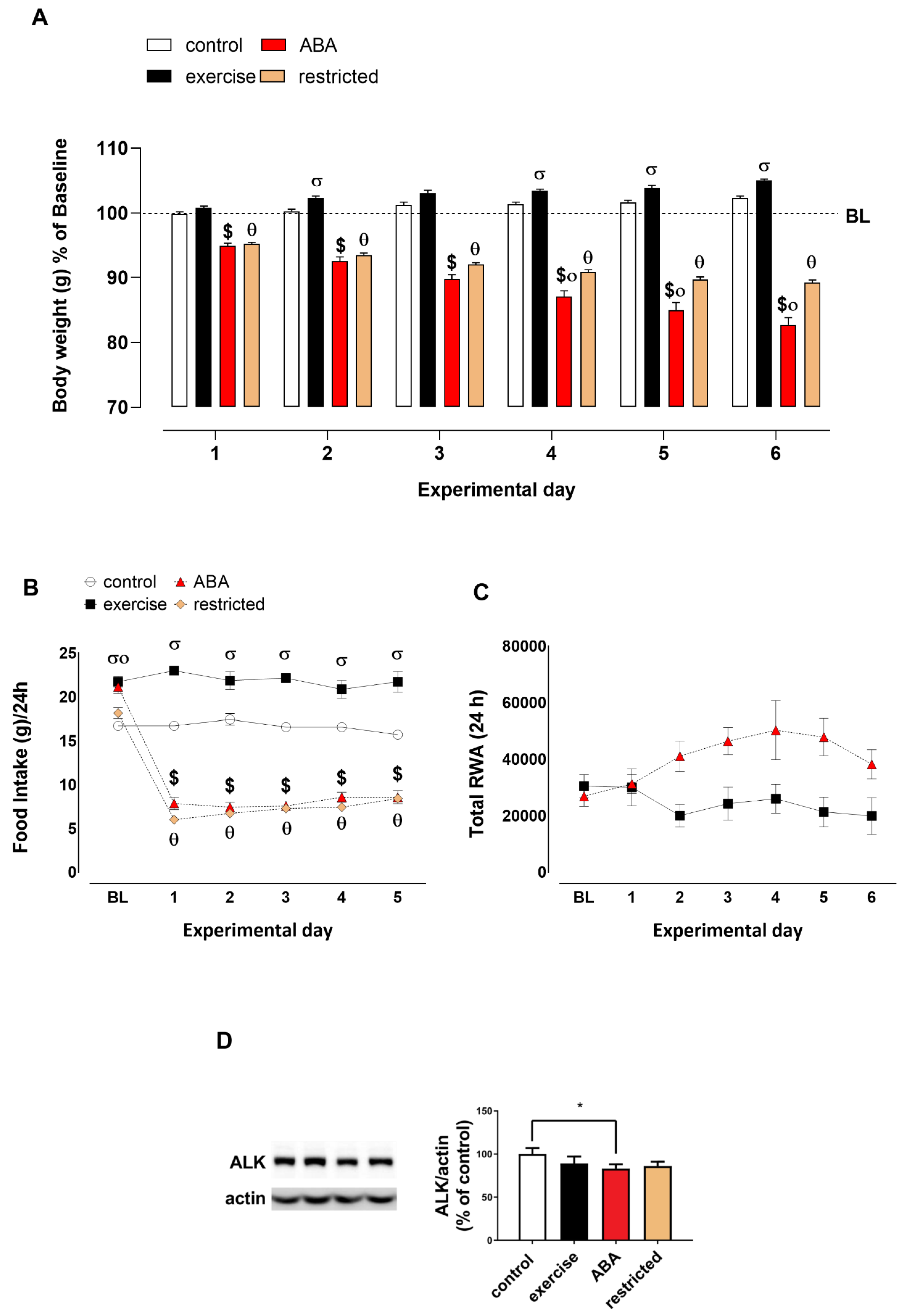
3.2. Recovery Phase
Body Weight, Food Intake, RWA, and ALK Receptor Expression
3.3. Second ABA Induction
Body Weight, Food Intake, RWA, and ALK Receptor Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoek, H.W. Incidence, prevalence and mortality of anorexia nervosa and other eating disorders. Curr. Opin. Psychiatry 2006, 19, 389–394. [Google Scholar] [CrossRef] [PubMed]
- van Eeden, A.E.; van Hoeken, D.; Hoek, H.W. Incidence, prevalence and mortality of anorexia nervosa and bulimia nervosa. Curr. Opin. Psychiatry 2021, 34, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Konstantynowicz, J.; Kadziela-Olech, H.; Kaczmarski, M.; Zebaze, R.M.; Iuliano-Burns, S.; Piotrowska-Jastrzebska, J.; Seeman, E. Depression in anorexia nervosa: A risk factor for osteoporosis. J. Clin. Endocrinol. Metab. 2005, 90, 5382–5385. [Google Scholar] [CrossRef]
- Gibson, D.; Watters, A.; Cost, J.; Mascolo, M.; Mehler, P.S. Extreme anorexia nervosa: Medical findings, outcomes, and inferences from a retrospective cohort. J. Eat. Disord. 2020, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Smink, F.R.; van Hoeken, D.; Hoek, H.W. Epidemiology of eating disorders: Incidence, prevalence and mortality rates. Curr. Psychiatry Rep. 2012, 14, 406–414. [Google Scholar] [CrossRef]
- Zipfel, S.; Giel, K.E.; Bulik, C.M.; Hay, P.; Schmidt, U. Anorexia nervosa: Aetiology, assessment, and treatment. Lancet Psychiatry 2015, 2, 1099–1111. [Google Scholar] [CrossRef]
- Steinhausen, H.C. The outcome of anorexia nervosa in the 20th century. Am. J. Psychiatry 2002, 159, 1284–1293. [Google Scholar] [CrossRef]
- Baenas, I.; Caravaca-Sanz, E.; Granero, R.; Sánchez, I.; Riesco, N.; Testa, G.; Vintró-Alcaraz, C.; Treasure, J.; Jiménez-Murcia, S.; Fernández-Aranda, F. COVID-19 and eating disorders during confinement: Analysis of factors associated with resilience and aggravation of symptoms. Eur. Eat. Disord. Rev. 2020, 28, 855–863. [Google Scholar] [CrossRef]
- Schlegl, S.; Maier, J.; Meule, A.; Voderholzer, U. Eating disorders in times of the COVID-19 pandemic-Results from an online survey of patients with anorexia nervosa. Int. J. Eat. Disord. 2020, 11, 1791–1800. [Google Scholar] [CrossRef]
- Termorshuizen, J.D.; Watson, H.J.; Thornton, L.M.; Borg, S.; Flatt, R.E.; MacDermod, C.M.; Harper, L.E.; van Furth, E.F.; Peat, C.M.; Bulik, C.M. Early impact of COVID-19 on individuals with self-reported eating disorders: A survey of ~1000 individuals in the United States and the Netherlands. Int. J. Eat. Disord. 2020, 53, 1780–1790. [Google Scholar] [CrossRef]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Créminon, C.; Vigny, M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef] [PubMed]
- Koyama-Nasu, R.; Haruta, R.; Nasu-Nishimura, Y.; Taniue, K.; Katou, Y.; Shirahige, K.; Todo, T.; Ino, Y.; Mukasa, A.; Saito, N.; et al. The pleiotrophin-ALK axis is required for tumorigenicity of glioblastoma stem cells. Oncogene 2014, 33, 2236–2244. [Google Scholar] [CrossRef]
- Murray, P.B.; Lax, I.; Reshetnyak, A.; Ligon, G.F.; Lillquist, J.S.; Natoli, E.J., Jr.; Shi, X.; Folta-Stogniew, E.; Gunel, M.; Alvarado, D.; et al. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci. Signal. 2015, 8, ra6. [Google Scholar] [CrossRef]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef]
- Cussac, D.; Greenland, C.; Roche, S.; Bai, R.Y.; Duyster, J.; Morris, S.W.; Delsol, G.; Allouche, M.; Payrastre, B. Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell lymphoma recruits, activates, and uses pp60c-src to mediate its mitogenicity. Blood 2004, 103, 1464–1471. [Google Scholar] [CrossRef]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef]
- Orthofer, M.; Valsesia, A.; Mägi, R.; Wang, Q.P.; Kaczanowska, J.; Kozieradzki, I.; Leopoldi, A.; Cikes, D.; Zopf, L.M.; Tretiakov, E.O.; et al. Identification of ALK in Thinness. Cell 2020, 181, 1246–1262. [Google Scholar] [CrossRef]
- Ahmed, M.; Kaur, N.; Cheng, Q.; Shanabrough, M.; Tretiakov, E.O.; Harkany, T.; Horvath, T.L.; Schlessinger, J. A hypothalamic pathway for Augmentor α-controlled body weight regulation. Proc. Natl. Acad. Sci. USA 2022, 119, e2200476119. [Google Scholar] [CrossRef] [PubMed]
- Scherma, M.; Collu, R.; Satta, V.; Giunti, E.; Fadda, P. Animal Models of Eating Disorders. Methods Mol. Biol. 2019, 2011, 297–314. [Google Scholar]
- Florent, V.; Baroncini, M.; Jissendi-Tchofo, P.; Lopes, R.; Vanhoutte, M.; Rasika, S.; Pruvo, J.P.; Vignau, J.; Verdun, S.; Johansen, J.E.; et al. Hypothalamic Structural and Functional Imbalances in Anorexia Nervosa. Neuroendocrinology 2020, 110, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.I.; Hiripi, E.; Pope, H.G., Jr.; Kessler, R.C. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol. Psychiatry 2007, 61, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. NC3Rs Reporting Guidelines Working Group. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef]
- Scherma, M.; Satta, V.; Collu, R.; Boi, M.F.; Usai, P.; Fratta, W.; Fadda, P. Cannabinoid CB1/CB2 receptor agonists attenuate hyperactivity and body weight loss in a rat model of activity-based anorexia. Br. J. Pharmacol. 2017, 174, 2682–2695. [Google Scholar] [CrossRef]
- Collu, R.; Scherma, M.; Piscitelli, F.; Giunti, E.; Satta, V.; Castelli, M.P.; Verde, R.; Fratta, W.; Bisogno, T.; Fadda, P. Impaired brain endocannabinoid tone in the activity-based model of anorexia nervosa. Int. J. Eat. Disord. 2019, 52, 1251–1262. [Google Scholar] [CrossRef]
- Ratnovsky, Y.; Neuman, P. The effect of pre-exposure and recovery type on activity-based anorexia in rats. Appetite 2011, 56, 567–576. [Google Scholar] [CrossRef]
- Dedoni, S.; Olianas, M.C.; Ingianni, A.; Onali, P. Type I interferons up-regulate the expression and signalling of p75 NTR/TrkA receptor complex in differentiated human SH-SY5Y neuroblastoma cells. Neuropharmacology 2014, 79, 321–334. [Google Scholar] [CrossRef]
- Dedoni, S.; Marras, L.; Olianas, M.C.; Ingianni, A.; Onali, P. Downregulation of TrkB Expression and Signaling by Valproic Acid and Other Histone Deacetylase Inhibitors. J. Pharmacol. Exp. Ther. 2019, 370, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Yano, W.; Kubota, T.; Yamauchi, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Okamoto, S.; Shiuchi, T.; et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007, 6, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, H.; Funahashi, T.; Kurokawa, N.; Sayama, K.; Fukuda, E.; Okita, K.; Imagawa, A.; Yamagata, K.; Shimomura, I.; Miyagawa, J.I.; et al. Plasma adiponectin levels in women with anorexia nervosa. Horm. Metab. Res. 2003, 35, 537–540. [Google Scholar]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar]
- Murugan, A.K.; Xing, M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011, 71, 4403–4411. [Google Scholar] [CrossRef]
- Souttou, B.; Carvalho, N.B.; Raulais, D.; Vigny, M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J. Biol. Chem. 2001, 276, 9526–9531. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–562. [Google Scholar] [CrossRef]
- Artmann, H.; Grau, H.; Adelmann, M.; Schleiffer, R. Reversible and non-reversible enlargement of cerebrospinal fluid spaces in anorexia nervosa. Neuroradiology 1985, 27, 304–312. [Google Scholar] [CrossRef]
- Docx, M.K.; Gewillig, M.; Simons, A.; Vandenberghe, P.; Weyler, J.; Ramet, J.; Mertens, L. Pericardial effusions in adolescent girls with anorexia nervosa: Clinical course and risk factors. Eat. Disord. 2010, 18, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Mourali, J.; Bénard, A.; Lourenço, F.C.; Monnet, C.; Greenland, C.; Moog-Lutz, C.; Racaud-Sultan, C.; Gonzalez-Dunia, D.; Vigny, M.; Mehlen, P.; et al. Anaplastic lymphoma kinase is a dependence receptor whose proapoptotic functions are activated by caspase cleavage. Mol. Cell Biol. 2006, 26, 6209–6222. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R.B.; El Hachem, C. Adiponectin in eating disorders. Eat. Weight Disord. 2014, 19, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 12, 988–1004. [Google Scholar] [CrossRef]
- Turnley, A.M.; Stapleton, D.; Mann, R.J.; Witters, L.A.; Kemp, B.E.; Bartlett, P.F. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J. Neurochem. 1999, 72, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Fraga, A.; Rial-Pensado, E.; Nogueiras, R.; Fernø, J.; Diéguez, C.; Gutierrez, E.; López, M. Activity-Based Anorexia Induces Browning of Adipose Tissue Independent of Hypothalamic AMPK. Front. Endocrinol. 2021, 12, 669980. [Google Scholar] [CrossRef]
- Iio, W.; Tokutake, Y.; Koike, H.; Matsukawa, N.; Tsukahara, T.; Chohnan, S.; Toyoda, A. Food Restriction and Malonyl-CoA. Anim. Sci. J. 2015, 86, 181–188. [Google Scholar] [CrossRef]
- Nobis, S.; Goichon, A.; Achamrah, N.; Guérin, C.; Azhar, S.; Chan, P.; Morin, A.; Bôle-Feysot, C.; do Rego, J.C.; Vaudry, D.; et al. Alterations of proteome, mitochondrial dynamic and autophagy in the hypothalamus during activity-based anorexia. Sci. Rep. 2018, 8, 7233. [Google Scholar] [CrossRef]
- Spadini, S.; Ferro, M.; Lamanna, J.; Malgaroli, A. Activity-based anorexia animal model: A review of the main neurobiological findings. J. Eat. Disord. 2021, 9, 123. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedoni, S.; Scherma, M.; Camoglio, C.; Siddi, C.; Fratta, W.; Fadda, P. Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa. Nutrients 2023, 15, 2205. https://doi.org/10.3390/nu15092205
Dedoni S, Scherma M, Camoglio C, Siddi C, Fratta W, Fadda P. Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa. Nutrients. 2023; 15(9):2205. https://doi.org/10.3390/nu15092205
Chicago/Turabian StyleDedoni, Simona, Maria Scherma, Chiara Camoglio, Carlotta Siddi, Walter Fratta, and Paola Fadda. 2023. "Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa" Nutrients 15, no. 9: 2205. https://doi.org/10.3390/nu15092205
APA StyleDedoni, S., Scherma, M., Camoglio, C., Siddi, C., Fratta, W., & Fadda, P. (2023). Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa. Nutrients, 15(9), 2205. https://doi.org/10.3390/nu15092205