Could Gut Microbiota Composition Be a Useful Indicator of a Long-Term Dietary Pattern?

 , , ,
, , ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Study Subjects
2.3. Anthropometric Measurements
2.4. Serum Biomarkers
2.5. Gut Microbiota Composition
2.6. Questionnaires
2.6.1. Lifestyle Questionnaire
2.6.2. Gastrointestinal Symptoms and Stool Consistency
2.6.3. Physical Activity
2.7. Dietary Intake and Adherence to Mediterranean Diet
2.8. Statistical Analysis
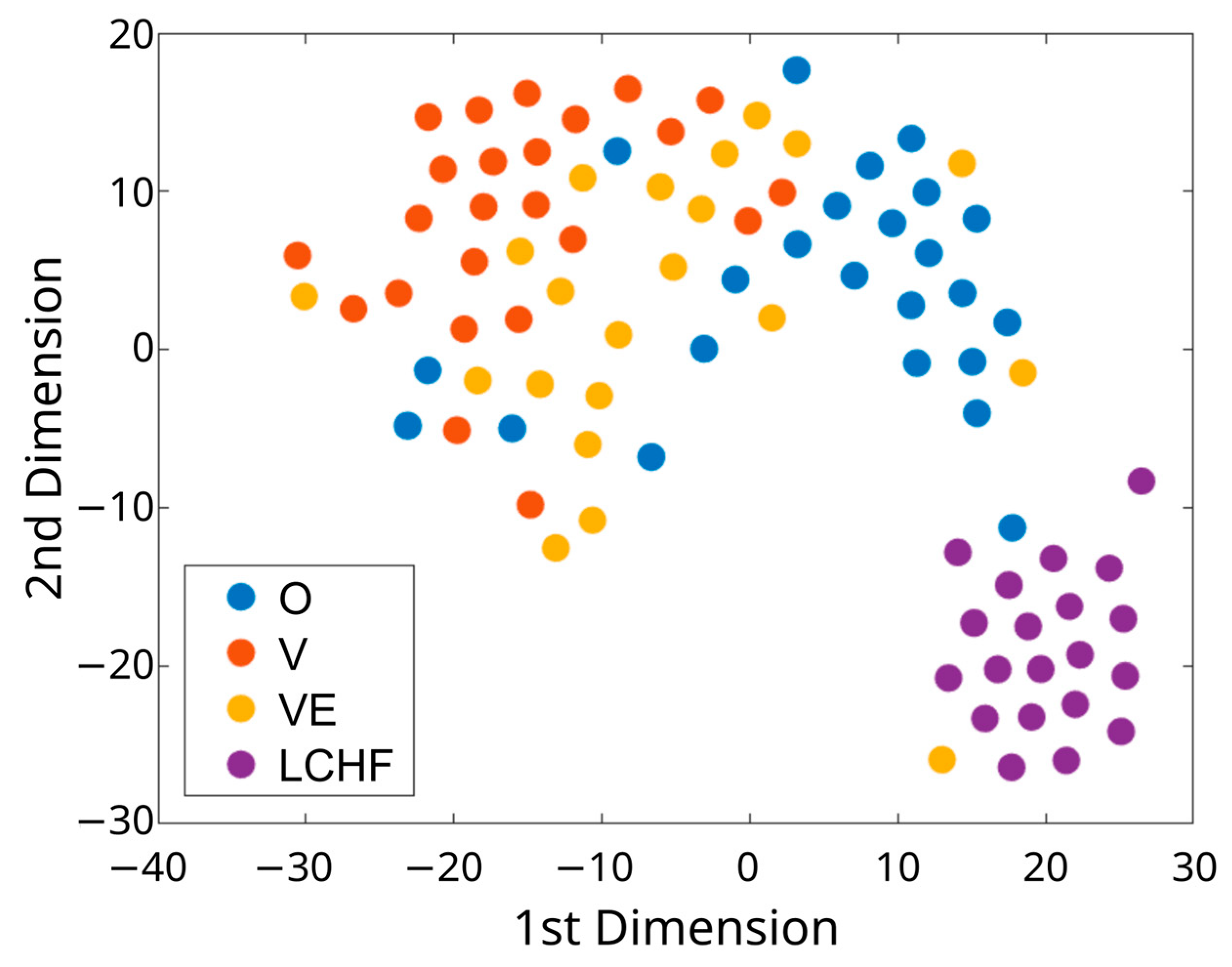
2.9. Visualization of High-Dimensional Dietary Data
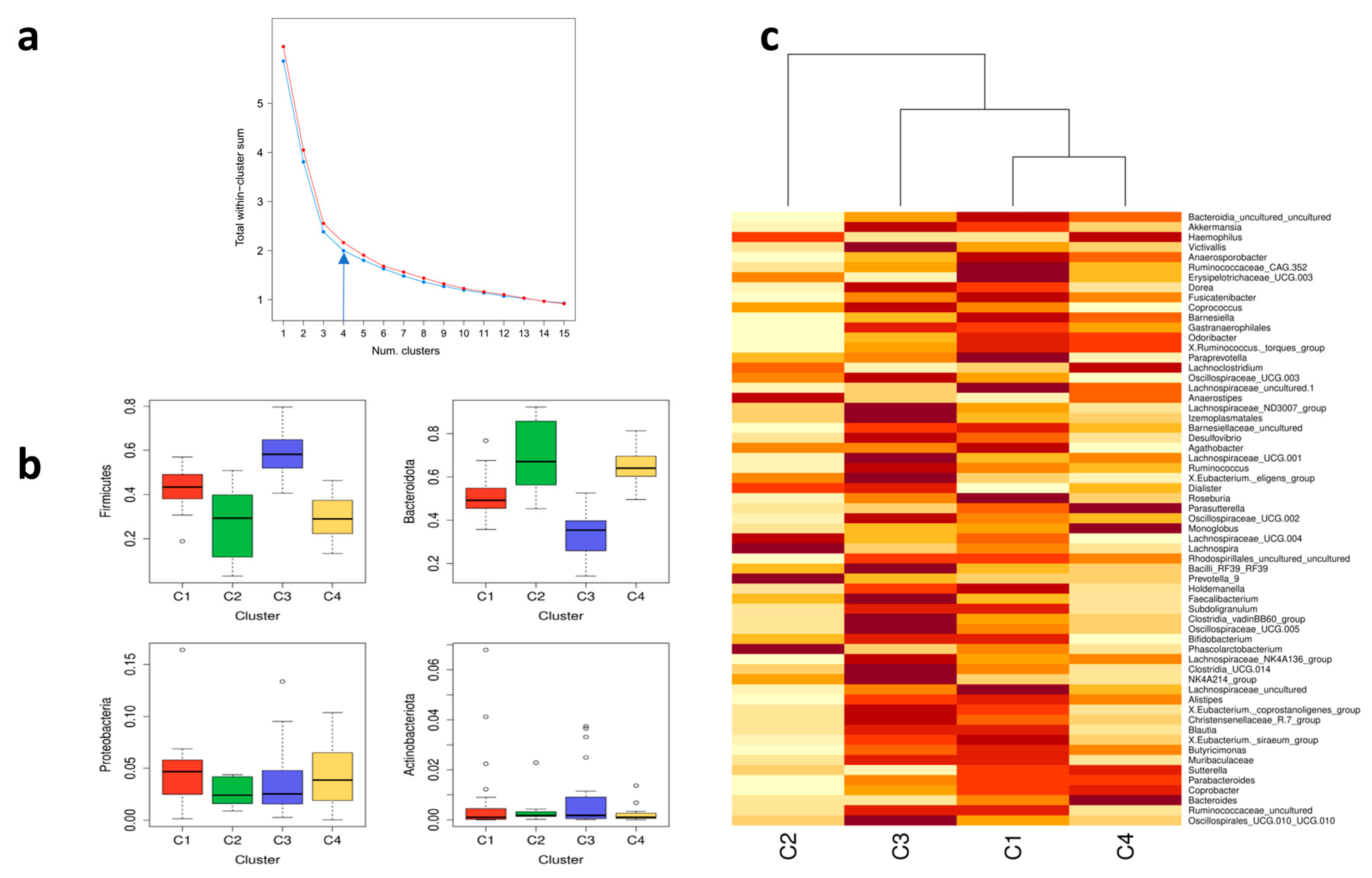
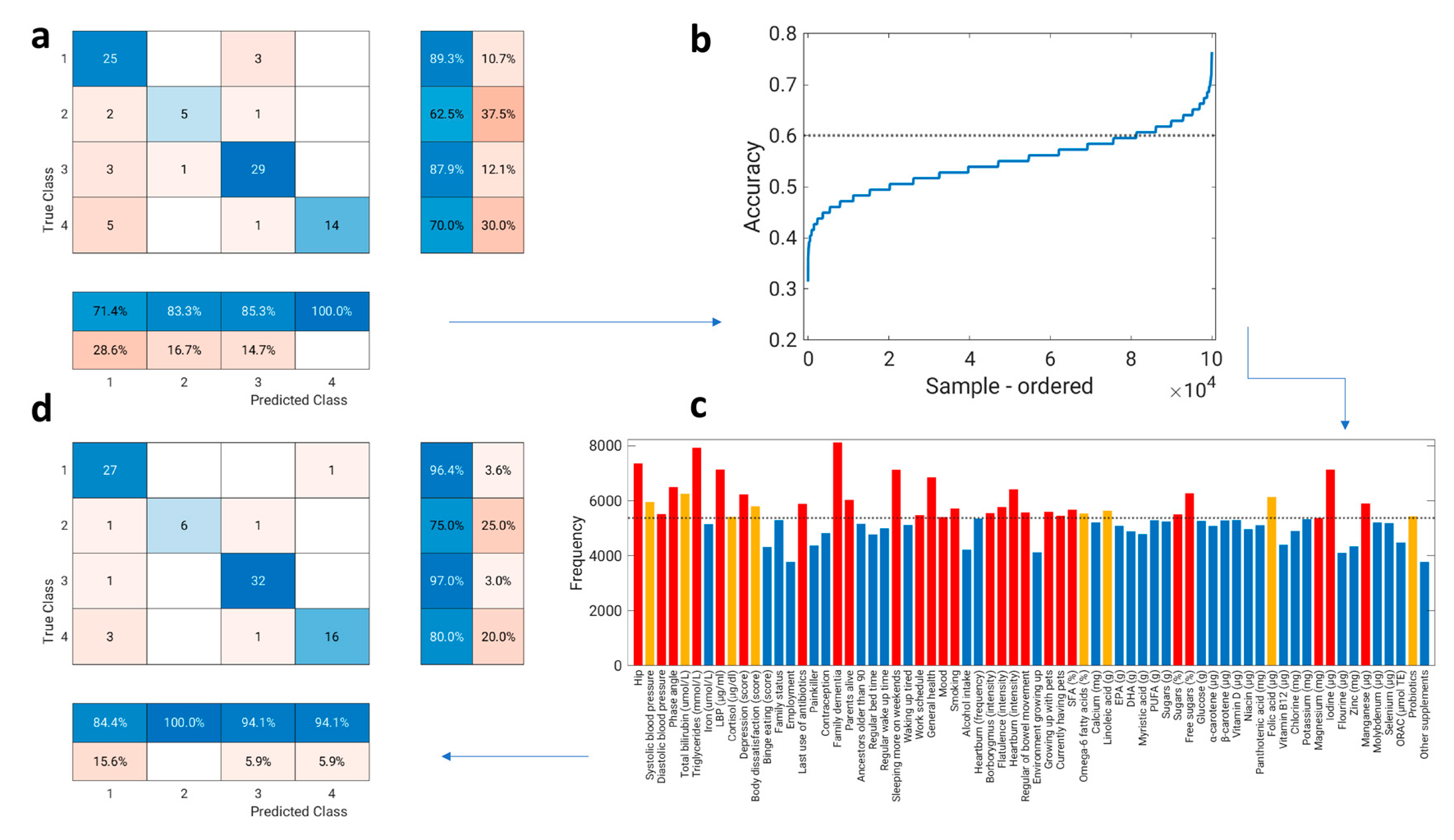
2.10. Cluster Analysis and General Predictors of Gut Microbiota Composition
3. Results
3.1. Characteristics of Subjects with Distinct Dietary Patterns
3.2. Serum Biomarkers in Subjects with Distinct Dietary Patterns
3.3. Dietary Intake in Subjects with Distinct Dietary Patterns
3.4. GI Symptoms and Gut Microbiota Composition in Subjects with Distinct Dietary Patterns
3.5. Cluster Analysis for Gut Microbiota Composition
3.6. Variable Selection
3.7. Predictors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, Y.; Pedersen, O. Gut Microbiota in Human Metabolic Health and Disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Kamiński, M.; Skonieczna-Żydecka, K.; Nowak, J.K.; Stachowska, E. Global and Local Diet Popularity Rankings, Their Secular Trends, and Seasonal Variation in Google Trends Data. Nutrition 2020, 79–80, 110759. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Ferrocino, I.; Di Cagno, R.; De Angelis, M.; Turroni, S.; Vannini, L.; Bancalari, E.; Rantsiou, K.; Cardinali, G.; Neviani, E.; Cocolin, L. Fecal Microbiota in Healthy Subjects Following Omnivore, Vegetarian and Vegan Diets: Culturable Populations and RRNA DGGE Profiling. PLoS ONE 2015, 10, e0128669. [Google Scholar] [CrossRef]
- Losasso, C.; Eckert, E.M.; Mastrorilli, E.; Villiger, J.; Mancin, M.; Patuzzi, I.; Di Cesare, A.; Cibin, V.; Barrucci, F.; Pernthaler, J.; et al. Assessing the Influence of Vegan, Vegetarian and Omnivore Oriented Westernized Dietary Styles on Human Gut Microbiota: A Cross Sectional Study. Front. Microbiol. 2018, 9, 317. [Google Scholar] [CrossRef]
- Franco-de-Moraes, A.C.; de Almeida-Pititto, B.; da Rocha Fernandes, G.; Gomes, E.P.; da Costa Pereira, A.; Ferreira, S.R.G. Worse Inflammatory Profile in Omnivores than in Vegetarians Associates with the Gut Microbiota Composition. Diabetol. Metab. Syndr. 2017, 9, 62. [Google Scholar] [CrossRef]
- Trefflich, I.; Jabakhanji, A.; Menzel, J.; Blaut, M.; Michalsen, A.; Lampen, A.; Abraham, K.; Weikert, C. Is a Vegan or a Vegetarian Diet Associated with the Microbiota Composition in the Gut? Results of a New Cross-Sectional Study and Systematic Review. Crit. Rev. Food Sci. Nutr. 2020, 60, 2990–3004. [Google Scholar] [CrossRef]
- Tyakht, A.V.; Alexeev, D.G.; Popenko, A.S.; Kostryukova, E.S.; Govorun, V.M. Rural and Urban Microbiota: To Be or Not to Be? Gut Microbes 2014, 5, 351–356. [Google Scholar] [CrossRef]
- Šik Novak, K.; Bogataj Jontez, N.; Kenig, S.; Hladnik, M.; Baruca Arbeiter, A.; Bandelj, D.; Černelič Bizjak, M.; Petelin, A.; Mohorko, N.; Jenko Pražnikar, Z. The Effect of COVID-19 Lockdown on Mental Health, Gut Microbiota Composition and Serum Cortisol Levels. Stress 2022, 25, 246–257. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A Communal Catalogue Reveals Earth’s Multiscale Microbial Diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible Sequence Taxonomy Reference Database Management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- Spielberger, C.; Gorsuch, R.; Lushene, R.; Vagg, P.; Jacobs, G. Manual for the State-Trait Anxiety Inventory (Form Y1–Y2); Consulting Psychologists Press: Palo Alto, CA, USA, 1983; Volume IV. [Google Scholar]
- Radloff, L.S. The CES-D Scale: A Self-Report Depression Scale for Research in the General Population. Appl. Psychol. Meas. 1977, 1, 385–401. [Google Scholar] [CrossRef]
- Watson, D.; Clark, L.A.; Tellegen, A. Development and Validation of Brief Measures of Positive and Negative Affect: The PANAS Scales. J. Personal. Soc. Psychol. 1988, 54, 1063–1070. [Google Scholar] [CrossRef]
- Donini, L.M.; Marsili, D.; Graziani, M.P.; Imbriale, M.; Cannella, C. Orthorexia Nervosa: Validation of a Diagnosis Questionnaire. Eat. Weight. Disord. 2005, 10, e28–e32. [Google Scholar] [CrossRef]
- Garner, D.M. Eating Disorder Inventory-2: Professional Manual; Psychological Assessment Resources: Odessa, FL, USA, 1991. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Publishing: Washington, DC, USA, 2013; ISBN 978-0-89042-557-2. [Google Scholar]
- Mason, T.B.; Lewis, R.J. Profiles of Binge Eating: The Interaction of Depressive Symptoms, Eating Styles, and Body Mass Index. Eat. Disord. 2014, 22, 450–460. [Google Scholar] [CrossRef]
- Revicki, D.A.; Wood, M.; Wiklund, I.; Crawley, J. Reliability and Validity of the Gastrointestinal Symptom Rating Scale in Patients with Gastroesophageal Reflux Disease. Qual. Life Res. 1998, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Heaton, K.W. Stool Form Scale as a Useful Guide to Intestinal Transit Time. Scand. J. Gastroenterol. 1997, 32, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Craig, C.L.; Marshall, A.L.; Sjöström, M.; Bauman, A.E.; Booth, M.L.; Ainsworth, B.E.; Pratt, M.; Ekelund, U.; Yngve, A.; Sallis, J.F.; et al. International Physical Activity Questionnaire: 12-Country Reliability and Validity. Med. Sci. Sport. Exerc. 2003, 35, 1381–1395. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, M.A.; García-Arellano, A.; Toledo, E.; Salas-Salvadó, J.; Buil-Cosiales, P.; Corella, D.; Covas, M.I.; Schröder, H.; Arós, F.; Gómez-Gracia, E.; et al. A 14-Item Mediterranean Diet Assessment Tool and Obesity Indexes among High-Risk Subjects: The PREDIMED Trial. PLoS ONE 2012, 7, e43134. [Google Scholar] [CrossRef]
- Redondo-Useros, N.; Nova, E.; González-Zancada, N.; Díaz, L.E.; Gómez-Martínez, S.; Marcos, A. Microbiota and Lifestyle: A Special Focus on Diet. Nutrients 2020, 12, 1776. [Google Scholar] [CrossRef]
- Heiss, S.; Hormes, J.M. Ethical Concerns Regarding Animal Use Mediate the Relationship between Variety of Pets Owned in Childhood and Vegetarianism in Adulthood. Appetite 2018, 123, 43–48. [Google Scholar] [CrossRef]
- Fresán, U.; Sabaté, J. Vegetarian Diets: Planetary Health and Its Alignment with Human Health. Adv. Nutr. 2019, 10, S380–S388. [Google Scholar] [CrossRef]
- Elliott, P.S.; Kharaty, S.S.; Phillips, C.M. Plant-Based Diets and Lipid, Lipoprotein, and Inflammatory Biomarkers of Cardiovascular Disease: A Review of Observational and Interventional Studies. Nutrients 2022, 14, 5371. [Google Scholar] [CrossRef]
- Burén, J.; Ericsson, M.; Damasceno, N.R.T.; Sjödin, A. A Ketogenic Low-Carbohydrate High-Fat Diet Increases LDL Cholesterol in Healthy, Young, Normal-Weight Women: A Randomized Controlled Feeding Trial. Nutrients 2021, 13, 814. [Google Scholar] [CrossRef]
- Mohorko, N.; Černelič-Bizjak, M.; Poklar-Vatovec, T.; Grom, G.; Kenig, S.; Petelin, A.; Jenko-Pražnikar, Z. Weight Loss, Improved Physical Performance, Cognitive Function, Eating Behavior, and Metabolic Profile in a 12-Week Ketogenic Diet in Obese Adults. Nutr. Res. 2019, 62, 64–77. [Google Scholar] [CrossRef]
- Retterstøl, K.; Svendsen, M.; Narverud, I.; Holven, K.B. Effect of Low Carbohydrate High Fat Diet on LDL Cholesterol and Gene Expression in Normal-Weight, Young Adults: A Randomized Controlled Study. Atherosclerosis 2018, 279, 52–61. [Google Scholar] [CrossRef]
- Gjuladin-Hellon, T.; Davies, I.G.; Penson, P.; Baghbadorani, R.A. Effects of Carbohydrate-Restricted Diets on Low-Density Lipoprotein Cholesterol Levels in Overweight and Obese Adults: A Systematic Review and Meta-Analysis. Nutr. Rev. 2019, 77, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, S.M.; Raposo, A.; Saraiva, A.; Zandonadi, R.P. Vegetarian Diet: An Overview through the Perspective of Quality of Life Domains. Int. J. Environ. Res. Public Health 2021, 18, 4067. [Google Scholar] [CrossRef]
- Yang, Q.; Lang, X.; Li, W.; Liang, Y. The Effects of Low-Fat, High-Carbohydrate Diets vs. Low-Carbohydrate, High-Fat Diets on Weight, Blood Pressure, Serum Liquids and Blood Glucose: A Systematic Review and Meta-Analysis. Eur. J. Clin. Nutr. 2022, 76, 16–27. [Google Scholar] [CrossRef]
- Bakaloudi, D.R.; Halloran, A.; Rippin, H.L.; Oikonomidou, A.C.; Dardavesis, T.I.; Williams, J.; Wickramasinghe, K.; Breda, J.; Chourdakis, M. Intake and Adequacy of the Vegan Diet. A Systematic Review of the Evidence. Clin. Nutr. 2021, 40, 3503–3521. [Google Scholar] [CrossRef]
- Clarys, P.; Deliens, T.; Huybrechts, I.; Deriemaeker, P.; Vanaelst, B.; De Keyzer, W.; Hebbelinck, M.; Mullie, P. Comparison of Nutritional Quality of the Vegan, Vegetarian, Semi-Vegetarian, Pesco-Vegetarian and Omnivorous Diet. Nutrients 2014, 6, 1318–1332. [Google Scholar] [CrossRef]
- Neufingerl, N.; Eilander, A. Nutrient Intake and Status in Adults Consuming Plant-Based Diets Compared to Meat-Eaters: A Systematic Review. Nutrients 2022, 14, 29. [Google Scholar] [CrossRef]
- Mooradian, A.D. The Merits and the Pitfalls of Low Carbohydrate Diet: A Concise Review. J. Nutr. Health Aging 2020, 24, 805–808. [Google Scholar] [CrossRef]
- Avital, K.; Buch, A.; Hollander, I.; Brickner, T.; Goldbourt, U. Adherence to a Mediterranean Diet by Vegetarians and Vegans as Compared to Omnivores. Int. J. Food Sci. Nutr. 2020, 71, 378–387. [Google Scholar] [CrossRef]
- Storz, M.A.; Rizzo, G.; Müller, A.; Lombardo, M. Bowel Health in U.S. Vegetarians: A 4-Year Data Report from the National Health and Nutrition Examination Survey (NHANES). Nutrients 2022, 14, 681. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, S.; Zhou, Y.; Yu, L.; Zhang, L.; Wang, Y. Altered Gut Microbiome Composition in Children with Refractory Epilepsy after Ketogenic Diet. Epilepsy Res. 2018, 145, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The Effects of Vegetarian and Vegan Diets on Gut Microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Matijašić, B.B.; Obermajer, T.; Lipoglavšek, L.; Grabnar, I.; Avguštin, G.; Rogelj, I. Association of Dietary Type with Fecal Microbiota in Vegetarians and Omnivores in Slovenia. Eur. J. Nutr. 2014, 53, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Prochazkova, M.; Budinska, E.; Kuzma, M.; Pelantova, H.; Hradecky, J.; Heczkova, M.; Daskova, N.; Bratova, M.; Modos, I.; Videnska, P.; et al. Vegan Diet Is Associated With Favorable Effects on the Metabolic Performance of Intestinal Microbiota: A Cross-Sectional Multi-Omics Study. Front. Nutr. 2022, 8, 783302. [Google Scholar] [CrossRef]
- Jaagura, M.; Viiard, E.; Karu-Lavits, K.; Adamberg, K. Low-Carbohydrate High-Fat Weight Reduction Diet Induces Changes in Human Gut Microbiota. MicrobiologyOpen 2021, 10, e1194. [Google Scholar] [CrossRef]
- Ma, W.; Nguyen, L.H.; Song, M.; Wang, D.D.; Franzosa, E.A.; Cao, Y.; Joshi, A.; Drew, D.A.; Mehta, R.; Ivey, K.L.; et al. Dietary Fiber Intake, the Gut Microbiome, and Chronic Systemic Inflammation in a Cohort of Adult Men. Genome Med. 2021, 13, 102. [Google Scholar] [CrossRef]
- Solopova, A.; van Sinderen, D. Determination of Bifidobacterial Carbohydrate Utilization Abilities and Associated Metabolic End Products. Methods Mol. Biol. 2021, 2278, 117–129. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Bailén, M.; Bressa, C.; Martínez-López, S.; González-Soltero, R.; Montalvo Lominchar, M.G.; San Juan, C.; Larrosa, M. Microbiota Features Associated With a High-Fat/Low-Fiber Diet in Healthy Adults. Front. Nutr. 2020, 7, 583608. [Google Scholar] [CrossRef]
- Kostovcikova, K.; Coufal, S.; Galanova, N.; Fajstova, A.; Hudcovic, T.; Kostovcik, M.; Prochazkova, P.; Jiraskova Zakostelska, Z.; Cermakova, M.; Sediva, B.; et al. Diet Rich in Animal Protein Promotes Pro-Inflammatory Macrophage Response and Exacerbates Colitis in Mice. Front. Immunol. 2019, 10, 919. [Google Scholar] [CrossRef] [PubMed]
- Shortt, C.; Hasselwander, O.; Meynier, A.; Nauta, A.; Fernández, E.N.; Putz, P.; Rowland, I.; Swann, J.; Türk, J.; Vermeiren, J.; et al. Systematic Review of the Effects of the Intestinal Microbiota on Selected Nutrients and Non-Nutrients. Eur. J. Nutr. 2018, 57, 25–49. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Gaundal, L.; Myhrstad, M.C.W.; Rud, I.; Gjøvaag, T.; Byfuglien, M.G.; Retterstøl, K.; Holven, K.B.; Ulven, S.M.; Telle-Hansen, V.H. Gut Microbiota Is Associated with Dietary Intake and Metabolic Markers in Healthy Individuals. Food Nutr. Res. 2022, 66, 8580. [Google Scholar] [CrossRef] [PubMed]
- Osborne, G.; Wu, F.; Yang, L.; Kelly, D.; Hu, J.; Li, H.; Jasmine, F.; Kibriya, M.G.; Parvez, F.; Shaheen, I.; et al. The Association between Gut Microbiome and Anthropometric Measurements in Bangladesh. Gut Microbes 2020, 11, 63–76. [Google Scholar] [CrossRef]
- Bezek, K.; Petelin, A.; Pražnikar, J.; Nova, E.; Redondo, N.; Marcos, A.; Jenko Pražnikar, Z. Obesity Measures and Dietary Parameters as Predictors of Gut Microbiota Phyla in Healthy Individuals. Nutrients 2020, 12, 2695. [Google Scholar] [CrossRef]
- Avery, E.G.; Bartolomaeus, H.; Maifeld, A.; Marko, L.; Wiig, H.; Wilck, N.; Rosshart, S.P.; Forslund, S.K.; Müller, D.N. The Gut Microbiome in Hypertension: Recent Advances and Future Perspectives. Circ. Res. 2021, 128, 934–950. [Google Scholar] [CrossRef]
- Sun, S.; Lulla, A.; Sioda, M.; Winglee, K.; Wu, M.C.; Jacobs, D.R.; Shikany, J.M.; Lloyd-Jones, D.M.; Launer, L.J.; Fodor, A.A.; et al. Gut Microbiota Composition and Blood Pressure. Hypertension 2019, 73, 998–1006. [Google Scholar] [CrossRef]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.M.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef]
- Mohr, A.E.; Crawford, M.; Jasbi, P.; Fessler, S.; Sweazea, K.L. Lipopolysaccharide and the Gut Microbiota: Considering Structural Variation. FEBS Lett. 2022, 596, 849–875. [Google Scholar] [CrossRef]
- Citronberg, J.S.; Curtis, K.R.; White, E.; Newcomb, P.A.; Newton, K.; Atkinson, C.; Song, X.; Lampe, J.W.; Hullar, M.A. Association of Gut Microbial Communities with Plasma Lipopolysaccharide-Binding Protein (LBP) in Premenopausal Women. ISME J. 2018, 12, 1631–1641. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. Biomed Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Moreno, M.; Perez-Herrera, A.; Locia-Morales, D.; Dizzel, S.; Meyre, D.; Stearns, J.C.; Cruz, M. Association of Gut Microbiome with Fasting Triglycerides, Fasting Insulin and Obesity Status in Mexican Children. Pediatr. Obes. 2021, 16, e12748. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef]
- Hung, C.-C.; Chang, C.-C.; Huang, C.-W.; Nouchi, R.; Cheng, C.-H. Gut Microbiota in Patients with Alzheimer’s Disease Spectrum: A Systematic Review and Meta-Analysis. Aging 2022, 14, 477–496. [Google Scholar] [CrossRef]
- Tun, H.M.; Konya, T.; Takaro, T.K.; Brook, J.R.; Chari, R.; Field, C.J.; Guttman, D.S.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; et al. Exposure to Household Furry Pets Influences the Gut Microbiota of Infants at 3–4 Months Following Various Birth Scenarios. Microbiome 2017, 5, 40. [Google Scholar] [CrossRef]
- Arenas-Montes, J.; Perez-Martinez, P.; Vals-Delgado, C.; Romero-Cabrera, J.L.; Cardelo, M.P.; Leon-Acuña, A.; Quintana-Navarro, G.M.; Alcala-Diaz, J.F.; Lopez-Miranda, J.; Camargo, A.; et al. Owning a Pet Is Associated with Changes in the Composition of Gut Microbiota and Could Influence the Risk of Metabolic Disorders in Humans. Animals 2021, 11, 2347. [Google Scholar] [CrossRef]
- Du, G.; Huang, H.; Zhu, Q.; Ying, L. Effects of Cat Ownership on the Gut Microbiota of Owners. PLoS ONE 2021, 16, e0253133. [Google Scholar] [CrossRef]
- Kates, A.E.; Jarrett, O.; Skarlupka, J.H.; Sethi, A.; Duster, M.; Watson, L.; Suen, G.; Poulsen, K.; Safdar, N. Household Pet Ownership and the Microbial Diversity of the Human Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 73. [Google Scholar] [CrossRef]
- Heddes, M.; Altaha, B.; Niu, Y.; Reitmeier, S.; Kleigrewe, K.; Haller, D.; Kiessling, S. The Intestinal Clock Drives the Microbiome to Maintain Gastrointestinal Homeostasis. Nat. Commun. 2022, 13, 6068. [Google Scholar] [CrossRef] [PubMed]
- Mortaş, H.; Bilici, S.; Karakan, T. The Circadian Disruption of Night Work Alters Gut Microbiota Consistent with Elevated Risk for Future Metabolic and Gastrointestinal Pathology. Chronobiol. Int. 2020, 37, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Antinozzi, M.; Giffi, M.; Sini, N.; Gallè, F.; Valeriani, F.; De Vito, C.; Liguori, G.; Romano Spica, V.; Cattaruzza, M.S. Cigarette Smoking and Human Gut Microbiota in Healthy Adults: A Systematic Review. Biomedicines 2022, 10, 510. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Ma, Z.; Jiao, M.; Wang, Y.; Li, A.; Ding, S. Effects of Smoking on Inflammatory Markers in a Healthy Population as Analyzed via the Gut Microbiota. Front. Cell. Infect. Microbiol. 2021, 11, 633242. [Google Scholar] [CrossRef]
- Elvers, K.T.; Wilson, V.J.; Hammond, A.; Duncan, L.; Huntley, A.L.; Hay, A.D.; van der Werf, E.T. Antibiotic-Induced Changes in the Human Gut Microbiota for the Most Commonly Prescribed Antibiotics in Primary Care in the UK: A Systematic Review. BMJ Open 2020, 10, e035677. [Google Scholar] [CrossRef]
- Ohkusa, T.; Koido, S.; Nishikawa, Y.; Sato, N. Gut Microbiota and Chronic Constipation: A Review and Update. Front. Med. 2019, 6, 19. [Google Scholar] [CrossRef]
- Mutuyemungu, E.; Singh, M.; Liu, S.; Rose, D.J. Intestinal Gas Production by the Gut Microbiota: A Review. J. Funct. Foods 2023, 100, 105367. [Google Scholar] [CrossRef]
- Manichanh, C.; Eck, A.; Varela, E.; Roca, J.; Clemente, J.C.; González, A.; Knights, D.; Knight, R.; Estrella, S.; Hernandez, C.; et al. Anal Gas Evacuation and Colonic Microbiota in Patients with Flatulence: Effect of Diet. Gut 2014, 63, 401–408. [Google Scholar] [CrossRef]
- Taylor, A.M.; Thompson, S.V.; Edwards, C.G.; Musaad, S.M.A.; Khan, N.A.; Holscher, H.D. Associations among Diet, the Gastrointestinal Microbiota, and Negative Emotional States in Adults. Nutr. Neurosci. 2020, 23, 983–992. [Google Scholar] [CrossRef]
- Simpson, C.A.; Diaz-Arteche, C.; Eliby, D.; Schwartz, O.S.; Simmons, J.G.; Cowan, C.S.M. The Gut Microbiota in Anxiety and Depression—A Systematic Review. Clin. Psychol. Rev. 2021, 83, 101943. [Google Scholar] [CrossRef]
- Winter, G.; Hart, R.A.; Charlesworth, R.P.G.; Sharpley, C.F. Gut Microbiome and Depression: What We Know and What We Need to Know. Rev. Neurosci. 2018, 29, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Sanada, K.; Nakajima, S.; Kurokawa, S.; Barceló-Soler, A.; Ikuse, D.; Hirata, A.; Yoshizawa, A.; Tomizawa, Y.; Salas-Valero, M.; Noda, Y.; et al. Gut Microbiota and Major Depressive Disorder: A Systematic Review and Meta-Analysis. J. Affect. Disord. 2020, 266, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Matsuda, K. How Microbes Affect Depression: Underlying Mechanisms via the Gut–Brain Axis and the Modulating Role of Probiotics. Int. J. Mol. Sci. 2022, 23, 1172. [Google Scholar] [CrossRef] [PubMed]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The Neuroactive Potential of the Human Gut Microbiota in Quality of Life and Depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-K.; Aring, L.; Das, N.K.; Solanki, S.; Inohara, N.; Iwase, S.; Samuelson, L.C.; Shah, Y.M.; Seo, Y.A. Impact of Dietary Manganese on Experimental Colitis in Mice. FASEB J. 2020, 34, 2929–2943. [Google Scholar] [CrossRef]
- Schiopu, C.; Ștefănescu, G.; Diaconescu, S.; Bălan, G.G.; Gimiga, N.; Rusu, E.; Moldovan, C.A.; Popa, B.; Tataranu, E.; Olteanu, A.V.; et al. Magnesium Orotate and the Microbiome–Gut–Brain Axis Modulation: New Approaches in Psychological Comorbidities of Gastrointestinal Functional Disorders. Nutrients 2022, 14, 1567. [Google Scholar] [CrossRef]
- Opazo, M.C.; Coronado-Arrázola, I.; Vallejos, O.P.; Moreno-Reyes, R.; Fardella, C.; Mosso, L.; Kalergis, A.M.; Bueno, S.M.; Riedel, C.A. The Impact of the Micronutrient Iodine in Health and Diseases. Crit. Rev. Food Sci. Nutr. 2022, 62, 1466–1479. [Google Scholar] [CrossRef]
- Wolters, M.; Ahrens, J.; Romaní-Pérez, M.; Watkins, C.; Sanz, Y.; Benítez-Páez, A.; Stanton, C.; Günther, K. Dietary Fat, the Gut Microbiota, and Metabolic Health—A Systematic Review Conducted within the MyNewGut Project. Clin. Nutr. 2019, 38, 2504–2520. [Google Scholar] [CrossRef]
- Satokari, R. High Intake of Sugar and the Balance between Pro- and Anti-Inflammatory Gut Bacteria. Nutrients 2020, 12, 1348. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O (n = 24) | V (n = 24) | VE (n = 21) | LCHF (n = 20) | ||
|---|---|---|---|---|---|
| Gender | % | % | % | % | p-value |
| Males/Females | 33.3/66.7 | 33.3/66.7 | 33.3/66.7 | 30.0/70.0 | 0.994 |
| Age | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Age (years) | 36.2 (10.4) | 33.6 (9.6) | 37.1 (10.8) | 39.4 (6.9) | 0.097 |
| Anthropometric measurements | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| BMI (kg/m2) | 22.2 (3.0) | 21.7 (2.2) | 22.3 (2.4) | 23.3 (3.2) | 0.381 |
| Waist circumference (cm) | 75.5 (9.8) | 74.5 (7.6) | 76.2 (8.8) | 76.8 (8.3) | 0.845 |
| Hip circumference (cm) | 95.5 (5.5) | 94.5 (5.1) | 96.2 (5.8) | 97.2 (7.1) | 0.477 |
| Fat mass (%) | 21.8 (7.3) | 19.7 (8.2) | 22.0 (7.2) | 21.5 (7.1) | 0.713 |
| Total body water (%) | 57.6 (6.0) | 58.7 (7.2) | 57.2 (6.1) | 57.9 (6.7) | 0.890 |
| Phase angle (°) | 6.6 (1.0) | 6.4 (0.9) | 6.2 (0.8) | 6.5 (1.0) | 0.559 |
| Blood pressure | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Systolic BP (mmHg) | 119.5 (13.6) | 124.2 (16.5) | 124.6 (18.3) | 121.8 (9.1) | 0.619 |
| Diastolic BP (mmHg) | 76.4 (8.8) | 77.1 (10.9) | 77.6 (11.8) | 77.4 (7.9) | 0.890 |
| Family status | % | % | % | % | p-value |
| Single | 29.2 | 29.2 | 19.0 | 10.0 | 0.367 |
| In a relationship or married | 70.8 | 70.8 | 81.0 | 90.0 | |
| Education | % | % | % | % | p-value |
| High school | 25.0 | 29.2 | 28.6 | 35.0 | 0.708 |
| Bachelor’s degree | 50.0 | 62.5 | 61.9 | 50.0 | |
| Master’s degree or PhD | 25.0 | 8.3 | 9.5 | 15.0 | |
| Socioeconomic status | % | % | % | % | p-value |
| Employed | 79.2 | 66.7 | 76.2 | 80.0 | 0.818 |
| Unemployed/housewife | 8.3 | 12.5 | 9.5 | 15.0 | |
| Student | 12.5 | 20.8 | 14.3 | 5.0 | |
| Work schedule | % | % | % | % | p-value |
| Not working | 4.2 | 12.5 | 19.0 | 10.0 | 0.225 |
| One-shift | 50.0 | 41.7 | 52.4 | 75.0 | |
| Two-shifts | 37.5 | 25.0 | 14.3 | 10.0 | |
| Flexible | 8.3 | 20.8 | 14.3 | 5.0 | |
| Living with | % | % | % | % | p-value |
| Alone | 16.7 | 20.8 | 14.3 | 5.0 | 0.220 |
| With partner and/or children | 62.5 | 54.2 | 61.9 | 85.0 | |
| With parents | 20.8 | 20.8 | 9.5 | 10.0 | |
| With friends/roommates | 0.0 | 4.2 | 14.3 | 0.0 | |
| Alcohol intake | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Alcohol (units/week) | 2.1 (3.8) | 1.0 (1.3) | 1.3 (1.8) | 2.0 (3.6) | 0.925 |
| Physical activity | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| IPAQ (MET/day) | 11.7 (9.3) | 11.8 (11.4) | 9.7 (8.7) | 7.5 (6.3) | 0.443 |
| Other lifestyle factors influencing gut microbiota | % | % | % | % | p-value |
| Smoking | 25.0 | 12.5 | 4.8 | 20.0 | 0.270 |
| Psychoactive substances use | 8.3 | 16.7 | 9.5 | 15.0 | 0.788 |
| Use of antibiotics in the last year | 20.9 | 25.0 | 0.0 | 5.0 | 0.104 |
| Allergies | 33.3 | 25.0 | 14.3 | 15.0 | 0.367 |
| Vaginal birth | 95.8 | 91.7 | 85.7 | 90.0 | 0.696 |
| Having been breastfed | 83.3 | 91.7 | 90.5 | 80.0 | 0.236 |
| Growing up in a rural environment | 29.2 | 75.0 | 61.9 | 60.0 | 0.012 * |
| Growing up with pets | 41.7 | 91.7 | 61.9 | 70.3 | 0.003 * |
| Currently living with pets | 54.2 | 54.2 | 57.1 | 55.0 | 0.997 |
| Serum Biomarkers | O (n = 24) | V (n = 24) | VE (n = 21) | LCHF (n = 20) | p-Value |
|---|---|---|---|---|---|
| M (SD) | M (SD) | M (SD) | M (SD) | ||
| Glucose (mmol/L) | 4.81 (0.48) | 4.63 (0.37) | 4.64 (0.48) | 4.63 (0.58) | 0.519 |
| Cholesterol (mmol/L) | 4.45 (0.72) | 4.00 (0.92) | 4.48 (0.88) | 7.57 (4.67) | <0.001 b,c,d |
| HDL (mmol/L) | 1.89 (0.45) | 1.58 (0.44) | 1.86 (0.38) | 2.16 (0.51) | 0.001 c |
| LDL (mmol/L) | 2.84 (0.64) | 2.67 (0.82) | 2.94 (0.86) | 5.87 (4.87) | <0.001 b,c,d |
| TAG (mmol/L) | 0.91 (0.61) | 0.88 (0.42) | 0.86 (0.32) | 0.92 (0.72) | 0.730 |
| Iron (μmol/L) | 27.79 (9.35) | 22.95 (10.00) | 18.11 (8.31) | 16.35 (6.38) | <0.001 a,b |
| AST (U/L) | 22.90 (10.22) | 21.54 (7.94) | 19.34 (6.44) | 18.94 (5.11) | 0.461 |
| Albumines (g/L) | 46.45 (4.63) | 46.15 (3.44) | 46.45 (3.52) | 45.39 (3.42) | 0.778 |
| Bilirubin (μmol/L) | 9.74 (5.68) | 8.88 (6.58) | 9.23 (4.17) | 6.77 (4.53) | 0.157 |
| CRP (mg/L) | 1.21 (2.01) | 0.57 (0.66) | 0.62 (0.80) | 0.73 (0.64) | 0.631 |
| LBP (μg/mL) | 4.31 (1.20) | 3.76 (1.60) | 3.57 (1.60) | 3.67 (1.95) | 0.608 |
| IL-6 (pg/mL) | 2.22 (2.80) | 1.45 (1.41) | 1.21 (0.59) | 2.47 (3.98) | 0.935 |
| TNF-α (pg/mL) | 0.45 (0.27) | 1.14 (2.99) | 0.78 (0.64) | 1.21 (1.93) | 0.149 |
| O (n = 24) | V (n = 24) | VE (n = 21) | LCHF (n = 20) | |||
|---|---|---|---|---|---|---|
| Energy | M (SD) | M (SD) | M (SD) | M (SD) | p-value | |
| Energy intake (kcal) | 2162 (800) | 2141 (716) | 2143 (664) | 1981 (568) | 0.895 | |
| Protein | M (SD) | M (SD) | M (SD) | M (SD) | p-value | RDI |
| Total protein (%) | 16.0 (3.1) | 12.3 (2.8) | 13.1 (3.7) | 22.5 (5.7) | <0.001 a,c,d,e | 10–15 |
| Plant protein (%) | 5.9 (2.1) | 11.6 (2.9) | 8.0 (2.3) | 1.9 (1.6) | <0.001 a,c,d,e | |
| Carbohydrates | M (SD) | M (SD) | M (SD) | M (SD) | p-value | RDI |
| Total carbohydrates (%) | 46.6 (7.1) | 59.0 (11.2) | 50.5 (10.4) | 9.4 (6.0) | <0.001 a,c,d,e | >50 |
| Sugars (%) | 16.3 (5.2) | 17.4 (8.3) | 17.7 (5.7) | 5.1 (3.9) | <0.001 c,d,e | |
| Free sugars (%) | 7.2 (4.0) | 4.1 (3.6) | 6.4 (2.6) | 1.6 (2.4) | <0.001 a,c,e | <10 |
| Dietary fiber (g) | 27.6 (17.6) | 55.4 (48.1) | 35.1 (13.9) | 16.3 (22.9) | <0.001 a,c,d,e | >30 |
| Fats | M (SD) | M (SD) | M (SD) | M (SD) | p-value | RDI |
| Total fats (%) | 35.5 (7.1) | 27.6 (9.8) | 36.1 (9.3) | 66.2 (8.2) | <0.001 a,c,d,e,f | 25–30 |
| SFA (%) | 10.7 (2.7) | 5.8 (3.2) | 9.8 (3.5) | 25.1 (6.1) | <0.001 a,c,d,e,f | <10 |
| MUFA (%) | 10.2 (3.8) | 9.6 (4.7) | 10.9 (5.1) | 22.0 (6.7) | <0.001 c,d,e | >10 |
| ω-3 PUFA (%) | 0.6 (0.6) | 0.7 (0.7) | 0.7 (0.7) | 1.1 (0.7) | 0.008 c,d | 0.5 |
| ω-6 PUFA (%) | 2.9 (2.1) | 3.1 (2.1) | 3.9 (2.6) | 4.6 (1.7) | 0.010 c | 2.5 |
| ω-3/ω-6 PUFA (ratio) | 0.2 (0.2) | 0.4 (0.7) | 0.2 (0.2) | 0.3 (0.2) | 0.333 | >0.2 |
| EPA (mg) | 131.7 (218.4) | 46.6 (152.7) | 72.5 (154.6) | 370.3 (533.5) | <0.001 a,b,d,e | |
| DHA (mg) | 268.9 (449.4) | 47.7 (129.3) | 104.8 (223.5) | 530.2 (675.9) | <0.001 a,b,d,e | |
| Cholesterol (mg) | 322.6 (181.8) | 12.4 (18.2) | 137.9 (126.1) | 1106.0 (529.9) | <0.001 a,c,d,e,f | |
| Use of probiotics | % | % | % | % | p-value | |
| Probiotics | 16.7 | 29.2 | 4.8 | 0.0 | 0.021 * | |
| Adherence to Mediterranean diet | M (SD) | M (SD) | M (SD) | M (SD) | p-value | |
| MEDAS (score) | 6.8 (2.3) | 8.8 (1.7) | 8.2 (1.8) | 5.9 (1.8) | <0.001 a,d,e |
| O (n = 24) | V (n = 24) | VE (n = 21) | LCHF (n = 20) | ||
|---|---|---|---|---|---|
| Stool consistency | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Bristol stool scale (score) | 3.9 (0.9) | 4.2 (1.2) | 3.8 (0.7) | 3.3 (1.0) | 0.019 b |
| GI symptoms | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Nausea (frequency) | 0.5 (0.8) | 0.4 (0.7) | 0.3 (0.7) | 0.2 (0.4) | 0.415 |
| Bloating (frequency) | 1.6 (1.1) | 1.7 (1.0) | 0.9 (0.9) | 1.0 (1.1) | 0.018 * |
| Borborygmi (frequency) | 1.3 (1.0) | 1.5 (1.1) | 1.1 (0.9) | 0.9 (1.0) | 0.186 |
| Abdominal pain (frequency) | 0.6 (0.7) | 0.8 (0.8) | 0.5 (0.8) | 0.4 (0.7) | 0.151 |
| Flatulence (frequency) | 1.7 (0.9) | 1.9 (0.9) | 1.5 (0.9) | 0.8 (0.9) | 0.002 a,b |
| Heartburn (frequency) | 0.5 (0.9) | 0.7 (1.0) | 0.6 (1.0) | 0.4 (0.6) | 0.763 |
| O (n = 24) | V (n = 24) | VE (n = 21) | LCHF (n = 20) | ||
| Phylum | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Firmicutes (%) | 45.41 (16.34) | 42.07 (16.33) | 47.13 (17.24) | 42.56 (15.19) | 0.702 |
| Bacteroidota (%) | 47.37 (17.64) | 51.67 (18.19) | 46.35 (16.61) | 51.74 (14.88) | 0.611 |
| Proteobacteria (%) | 4.19 (2.15) | 4.06 (3.18) | 4.34 (3.79) | 3.61 (2.58) | 0.691 |
| Verrucomicrobiota (%) | 1.02 (1.12) | 0.36 (0.76) | 0.69 (0.89) | 0.51 (0.65) | 0.020 a |
| Cyanobacteria (%) | 1.00 (3.00) | 0.45 (0.84) | 0.91 (1.60) | 0.84 (1.69) | 0.577 |
| Actinobacteria (%) | 0.79 (1.11) | 0.96 (1.64) | 0.27 (0.28) | 0.05 (0.06) | <0.001 b,c,d |
| Desulfobacterota (%) | 0.21 (0.23) | 0.37 (0.67) | 0.32 (0.54) | 0.66 (0.67) | 0.018 c,d |
| Gut microbiota α-diversity | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Shannon index | 3.27 (0.48) | 2.85 (0.73) | 3.27 (0.49) | 3.33 (0.46) | 0.090 |
| Predictor | C1 (n = 28) | C2 (n = 8) | C3 (n = 33) | C4 (n = 20) | |
|---|---|---|---|---|---|
| Anthropometric measurements | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Hip circumference (cm) | 98.3 (5.9) | 94.3 (4.9) | 95.7 (5.6) | 92.8 (5.1) | 0.008 b |
| Phase angle (°) | 6.6 (0.9) | 7.2 (0.7) | 6.3 (0.8) | 6.3 (0.9) | 0.033 c |
| Diastolic blood pressure (mm Hg) | 77.1 (8.9) | 73.9 (5.8) | 78.8 (11.3) | 75.6 (9.8) | 0.747 |
| Serum biomarkers | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Serum TAG (mmol/L) | 1.10 (0.80) | 0.79 (0.24) | 0.72 (0.26) | 0.93 (0.36) | 0.092 |
| Serum LBP (μg/mL) | 4.24 (1.61) | 3.97 (1.46) | 3.58 (1.76) | 3.68 (1.29) | 0.500 |
| Subject’s lifestyle factors | % | % | % | % | p-value |
| Growing up with pets | 78.6 | 100.0 | 48.5 | 65.0 | 0.014 * |
| Currently having pets | 53.6 | 37.5 | 60.6 | 55.0 | 0.698 |
| Regular bowel movement | 89.3 | 87.5 | 69.7 | 75.0 | 0.262 |
| Smoking | 17.9 | 25.0 | 9.1 | 20.0 | 0.575 |
| Family history of dementia | 14.3 | 37.5 | 9.1 | 35.1 | 0.055 |
| Sleeping more on weekends | 57.1 | 37.5 | 63.6 | 55.0 | 0.600 |
| Work schedule (not working/one shift/two shifts/flexible) | 10.7/50.0/32.1/7.1 | 25.0/12.5/15.5/50.0 | 3.0/69.7/18.2/9.1 | 20.0/50.0/20.0/10.0 | 0.013 * |
| Last use of antibiotics (>1 year ago or never/6–12 months ago/3–5 months ago) | 92.9/0.0/7.1 | 62.5/37.5/0.0 | 87.9/9.1/3.0 | 85.0/10.0/5.0 | 0.076 |
| Parents alive (both/one/no one) | 78.6/7.1/14.3 | 100.0/0.0/0.0 | 72.7/27.3/0.0 | 90.0/5.0/5.0 | 0.024 * |
| GI symptoms | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Borborygmi (intensity) | 0.6 (0.7) | 0.9 (0.6) | 0.9 (0.8) | 1.0 (0.8) | 0.281 |
| Flatulence (intensity) | 1.2 (0.8) | 1.0 (0.8) | 1.2 (0.8) | 1.4 (0.8) | 0.694 |
| Heartburn (intensity) | 0.3 (0.5) | 0.6 (1.1) | 0.5 (0.7) | 0.6 (0.8) | 0.503 |
| Psychological factors | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| Subjective general health (score) | 4.6 (0.6) | 4.1 (0.4) | 4.3 (0.6) | 4.4 (0.7) | 0.172 |
| Subjective mood (score) | 4.2 (0.9) | 3.8 (0.7) | 4.0 (0.9) | 3.7 (0.8) | 0.062 |
| Symptoms of depression (score) | 8.5 (9.4) | 9.9 (8.0) | 7.7 (5.3) | 10.3 (5.6) | 0.206 |
| Specific nutrients intake | M (SD) | M (SD) | M (SD) | M (SD) | p-value |
| SFA (%) | 13.5 (9.0) | 5.2 (2.3) | 12.1 (6.5) | 14.2 (9.5) | 0.008 a,c,d |
| Sugars (%) | 14.1 (9.6) | 12.6 (4.1) | 14.1 (6.7) | 16.2 (8.2) | 0.421 |
| Free sugars (%) | 4.3 (3.6) | 3.4 (2.1) | 4.9 (3.5) | 6.3 (5.1) | 0.429 |
| Magnesium (mg) | 708.8 (673.3) | 695.6 (425.2) | 515.4 (479.2) | 415.7 (217.6) | 0.101 |
| Iodine (µg) | 78.5 (42.9) | 43.4 (27.2) | 113.3 (68.9) | 96.2 (47.5) | 0.006 c,d |
| Manganese (mg) | 14.5 (35.5) | 9.4 (6.6) | 8.3 (12.3) | 5.1 (3.4) | 0.144 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šik Novak, K.; Bogataj Jontez, N.; Petelin, A.; Hladnik, M.; Baruca Arbeiter, A.; Bandelj, D.; Pražnikar, J.; Kenig, S.; Mohorko, N.; Jenko Pražnikar, Z. Could Gut Microbiota Composition Be a Useful Indicator of a Long-Term Dietary Pattern? Nutrients 2023, 15, 2196. https://doi.org/10.3390/nu15092196
Šik Novak K, Bogataj Jontez N, Petelin A, Hladnik M, Baruca Arbeiter A, Bandelj D, Pražnikar J, Kenig S, Mohorko N, Jenko Pražnikar Z. Could Gut Microbiota Composition Be a Useful Indicator of a Long-Term Dietary Pattern? Nutrients. 2023; 15(9):2196. https://doi.org/10.3390/nu15092196
Chicago/Turabian StyleŠik Novak, Karin, Nives Bogataj Jontez, Ana Petelin, Matjaž Hladnik, Alenka Baruca Arbeiter, Dunja Bandelj, Jure Pražnikar, Saša Kenig, Nina Mohorko, and Zala Jenko Pražnikar. 2023. "Could Gut Microbiota Composition Be a Useful Indicator of a Long-Term Dietary Pattern?" Nutrients 15, no. 9: 2196. https://doi.org/10.3390/nu15092196
APA StyleŠik Novak, K., Bogataj Jontez, N., Petelin, A., Hladnik, M., Baruca Arbeiter, A., Bandelj, D., Pražnikar, J., Kenig, S., Mohorko, N., & Jenko Pražnikar, Z. (2023). Could Gut Microbiota Composition Be a Useful Indicator of a Long-Term Dietary Pattern? Nutrients, 15(9), 2196. https://doi.org/10.3390/nu15092196