
Phenylalanine Tolerance over Time in Phenylketonuria: A Systematic Review and Meta-Analysis
 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Literature Search
2.2. Study Selection
2.3. Outcome Measures
2.4. Data Extraction
2.5. Quality Appraisal and Risk of Bias Assessment
2.6. Data Analysis
3. Results
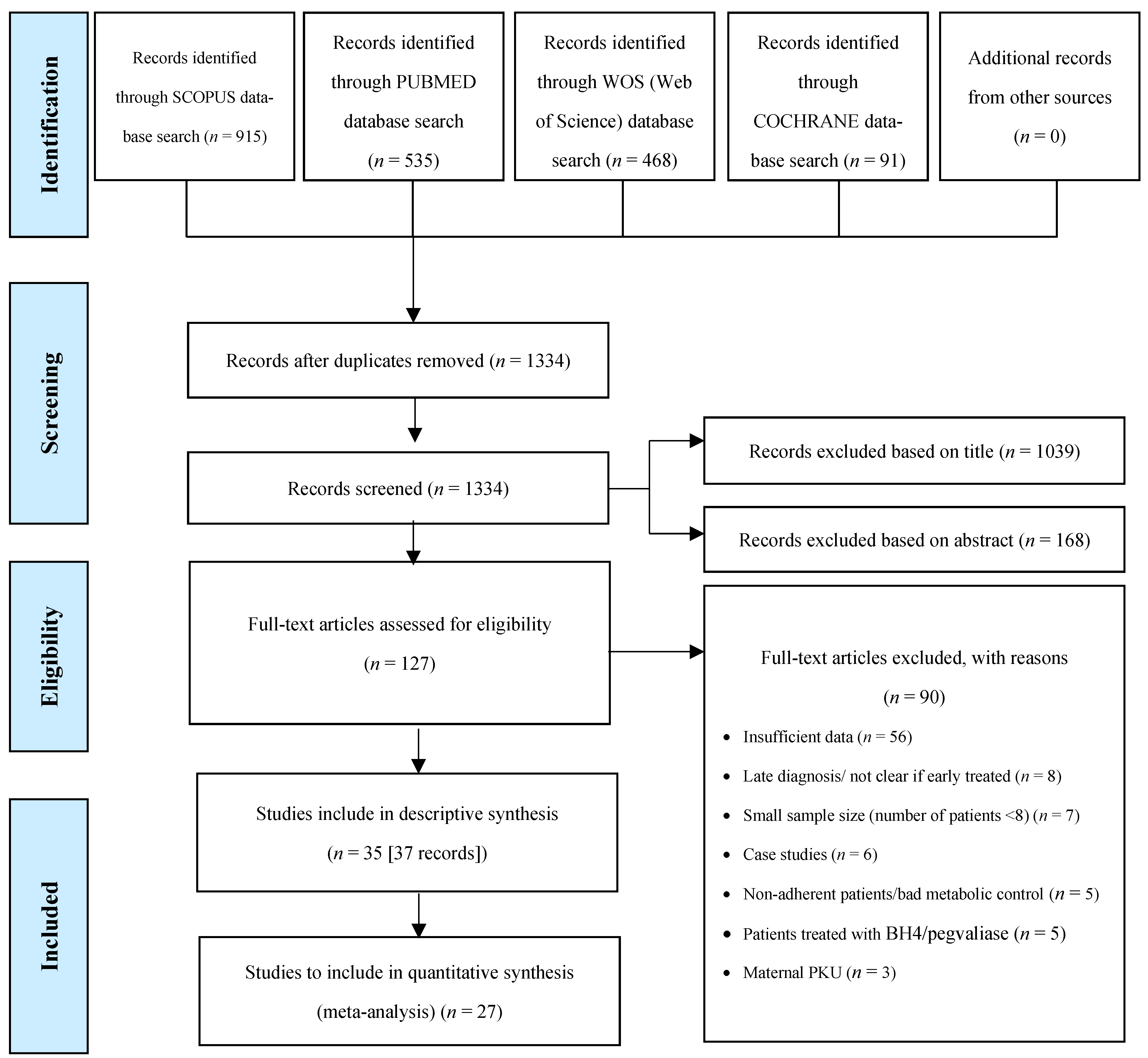
3.1. Study Selection
3.2. Study Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Country, Setting | Study Design | PKU Phenotype | Sample Size | Age | Gender (M:F) | Ethnic Origin | |
|---|---|---|---|---|---|---|---|---|
| Mean (SD) | Median (Range) | |||||||
| Acosta et al., 1998 [31] | USA, Multicentre | Prospective, longitudinal | cPKU | 35 | 13.7 (1.9) days | N/A | 20:15 | 33 Caucasian; 2 unknown |
| Aldámiz-Echevarría et al., 2014 [32] | Spain, 14 centres | Retrospective; longitudinal; multicenter | mildHPA mPKU moPKU cPKU All types | 226 43 78 158 505 a | N/A | (1–18 years) | 236:269 | Caucasian |
| Aldámiz-Echevarría et al., 2013 [33] | Spain, 13 centres | Retrospective; longitudinal | mPKU moPKU cPKU All types | 24 99 15 138 b | Pre-sapropterin Baseline: 2-y FU: 5.0 y (4.6) 5-y FU: 5.2 y (3.1) Diet Baseline: 2-y FU: 5.0 y 5-y FU: 5.0 y Diet Final: 2-y FU: 7.0 y 5-y FU: 10.2 (3.0) y | N/A | Pre-sapropterin Baseline: 2-y FU 18:18 5-y FU 6:4 Diet Group: 2-y FU 36:36 5-y FU 12:8 | Caucasian |
| Aldámiz-Echevarría et al., 2015 [34] | Spain, 14 centres | Retrospective; longitudinal | mPKU moPKU cPKU All types | 15 42 9 66 c | Pre-sapropterin Group: 16.9 (10.4) y Diet Group: 16.9 (10.3) y | (0–4 y) | Pre-sapropterin Group: 12:10 Diet Group: 24:20 | Caucasian |
| Alm et al., 1986 [27] | Sweden, single centre | Prospective; longitudinal | All types | 23 d | N/A | Final age: (8–19 y) | 12:11 | 22 Caucasian; 1 Syrian |
| Daly et al., 2017 [35] | UK, single centre | Prospective; longitudinal | All types | 22 e | N/A | 11 y (6–16 y) | 13:9 | 19 white European; 3 Pakistani |
| Daly et al., 2019 [36] | UK, 2 centres | Randomised; controlled; crossover | All types | 19 f | N/A | 10 y (6–16 y) | 7:11 | 17 white European; 2 Pakistani |
| Dobbelaere et al., 2003 [37] | France, single centre | Cross-sectional; longitudinal | All types | 20 | 4.5 (1.6) y | (8 mo–7 y) | 9:11 | N/A |
| Evans et al., 2017 [38] | Australia, single centre | Prospective; longitudinal | All types | 32 | 9.2 (4.7) y | (0.8–18 y) | 10:22 | N/A |
| Evans et al., 2018 [53] | UK, single centre | Retrospective; longitudinal | All types | 31 | N/A | Baseline: 20 wk (13–37 wk) Total: (3–24 mo) | 16:15 | N/A |
| Evans et al., 2019 [39] | UK, 3 centres | Open label; longitudinal; prospective; case–control | All types | 20 | N/A | Baseline: 4.3 mo (2.9–6.6 mo) Total: (3–24 mo) | 14:6 | Caucasian |
| Evers et al., 2018 [28] | The Netherlands 2 centres | Retrospective; cohort | All types | 39 g | Pre-sapropterin Group: 13.1 (9.2) y Diet Group: 13.0 (9.2) y | Pre-sapropterin Group: (2.8–33.7 y) Diet Group: (1.4–33.4 y) | Pre-sapropterin Group: 5:13 Diet Group: 7:14 | N/A |
| Ferguson et al., 1996 [54] | UK, Multicentre | Randomised controlled trial | All types | 20 h | N/A | (9–15 y) | 13:7 | N/A |
| Giovaninni et al., 2014 [40] | Italy, 1 centre | Randomised-controlled | mildHPA mPKU cPKU All types | 60 40 15 115 | mHPA: 9.3 (3.3) y cPKU + mPKU: 9.2 (3.4) y | N/A | 50:65 | N/A |
| Gökmen-Özel et al., 2011 [41] | UK, 2 centres | Randomised-controlled; crossover | All types | 14 | N/A | 6.3 (3.0–9.7) y | 12:2 | 13 Caucasian; 1 Asian |
| Green et al.; 2019 [42] | UK, Multicentre | Cross-sectional (pooled analysis of 2 multicenter intervention studies) | All types | 16 | 29.5 (11.2) y | N/A | 7:9 | N/A |
| Hoeksma et al., 2005 [43] | The Netherlands, 8 centres | Retrospective; longitudinal | All types | 174 i | N/A | (0–3 y) | N/A | Caucasian |
| Huemer et al., 2007 [44] | Austria, single centre | Prospective; longitudinal | All types | 34 | Baseline: 8.7 (3.9) y | (2 mo–15 y) | 22:12 | N/A |
| Kindt et al., 1983 [45] | Norway n/a | Prospective; longitudinal | All types | 16 | N/A | End of the study: (2–6 y) | 7:9 | N/A |
| MacDonald et al., 2006 [46] | UK, single centre | Randomised; prospective; crossover | All types | 25 | 6.0 (2.5) y | 6 (2–10) y | 11:14 | N/A |
| MacDonald et al.; 2003 [18] | UK, single centre | Randomised; crossover | All types | 16 j | 5.3 (3.1) y | 4.5 (2–11) y | 4:12 | 15 Caucasian; 1 mixed ethnicity |
| MacDonald et al., 1996 [57] | UK, single centre | Longitudinal observational study | All types | 19 | 6.6 (4.9) y | 4 (1–16) y | 15:4 | Caucasian |
| Pinto et al., 2019 [29] | Portugal, single centre | Retrospective; longitudinal | mildHPA mPKU cPKU All types | 10 23 7 40 | 17 y | (12–29 y) | 24:16 | Caucasian |
| Ponzone et al., 2008 [47] | Italy, single centre | Retrospective; longitudinal | mildHPA mPKU moPKU cPKU All types | 5 6 4 7 22 | 8.5 (4.3) y | 8 (1.7–14.7) y | 10:12 | N/A |
| Rocha et al., 2012 & 2013 [25,26] | Portugal, single centre | Cross-sectional | mildHPA mPKU cPKU All types | 18 42 29 89 | 14.4 (6.6) y | (3–30 y) | 48:41 | Caucasian |
| Rohde et al., 2012 & 2014a [19,20] | Germany, single centre | Randomised; cross-over | All types | 19 | 4.7 (2.1) y | (2–10) y | N/A | N/A |
| Rohde et al., 2015 [48] | Germany 10 centres | Retrospective; cross-sectional | All types | 149 | 7 (6.6) y | 7 (1–15) y | 77:72 | N/A |
| Rohde et al., 2014b [49] | Germany, Multicentre | Cross-sectional | All types | 67 | N/A | (6–45) y | N/A | N/A |
| Schulpis et al., 2013 [50] | Greece, single centre | Prospective; cohort | All types | 30 | 5.0 (3.2) y | N/A | 15:15 | N/A |
| Stockler-Ipsiroglu et al., 2015 [58] | Canada, single centre | Retrospective chart review | mildHPA mPKU moPKU cPKU All types | 4 1 1 5 11 | 5.4 (4.8) y | 4.5 y (1 mo–15.5 y) | 7:4 | N/A |
| Sweeney et al., 2012 [30] | Australia, single centre | Phase I: Randomised controlled Phase II: Prospective cohort | moPKU cPKU All types | 2 17 19 | 10.5 (5.7) y | 9.5 y (1.5–20.5) y | 6:13 | N/A |
| Thiele et al., 2017 [51] | Germany, 2 centres | Retrospective; longitudinal | mildHPA cPKU All types | 41 183 224 | N/A | (0–18 y) | 119:105 | Caucasian |
| Trefz et al., 2009 [52] | Germany, Poland, Spain, USA, 15 centres | International; double-blind randomised; placebo controlled | All types | 45 k | Pre-sapropterin Baseline: 7.7 (2.8) y Placebo Baseline: 7.1 (2.0) y Total: 7.4 y | (4–12 y) | Pre-sapropterin Group: 20:13 Placebo: 6:6 | N/A |
| van Spronsen et al., 2009 [55] | The Netherlands, 8 centres | Retrospective; longitudinal | All types | 213 | N/A | (1 mo–10 y) | N/A | N/A |
| Wendel et al., 1990 [56] | Germany, single centre | Retrospective; longitudinal | All types | 139 | N/A | (1–6 y) | 66:73 | N/A |
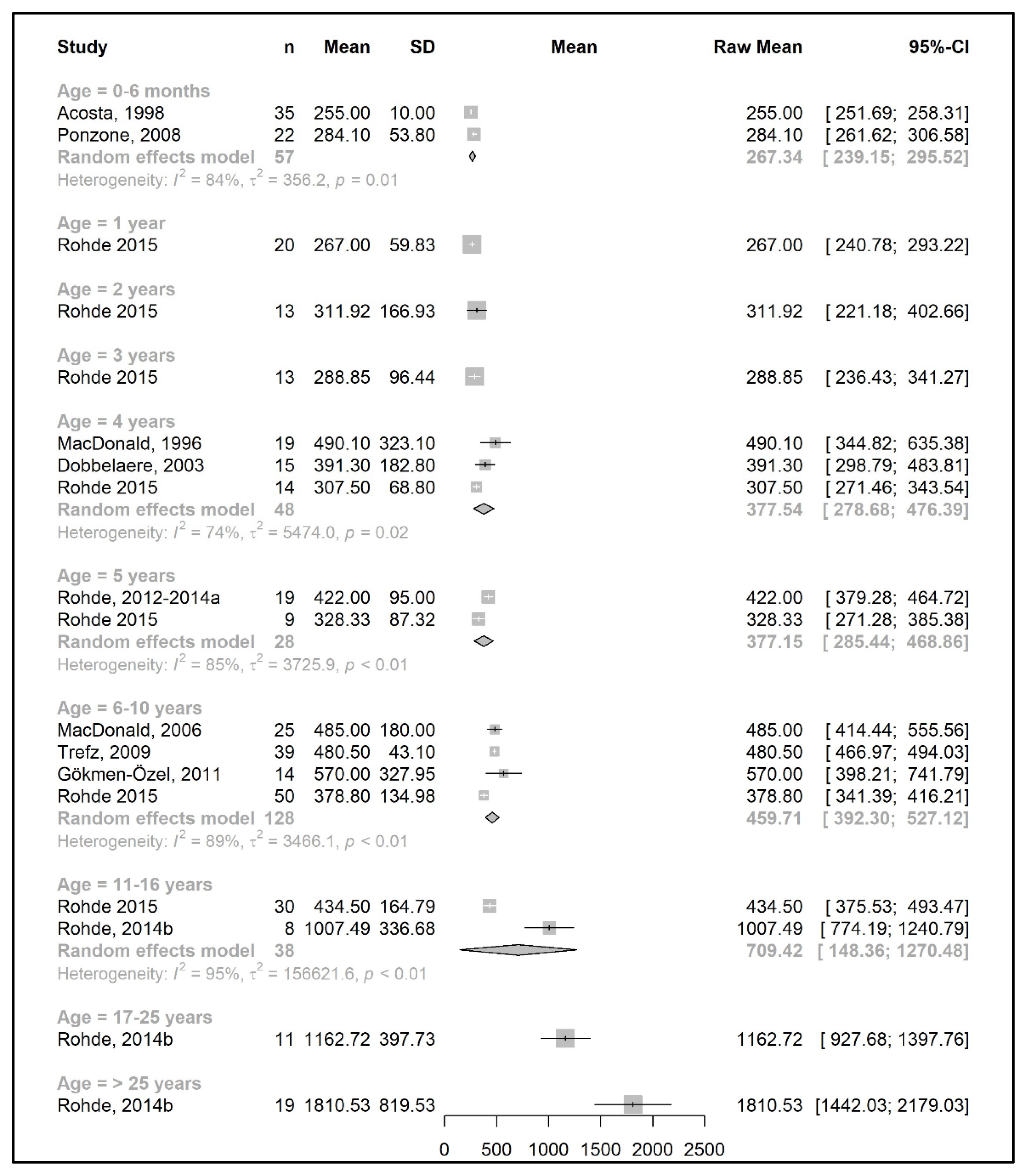
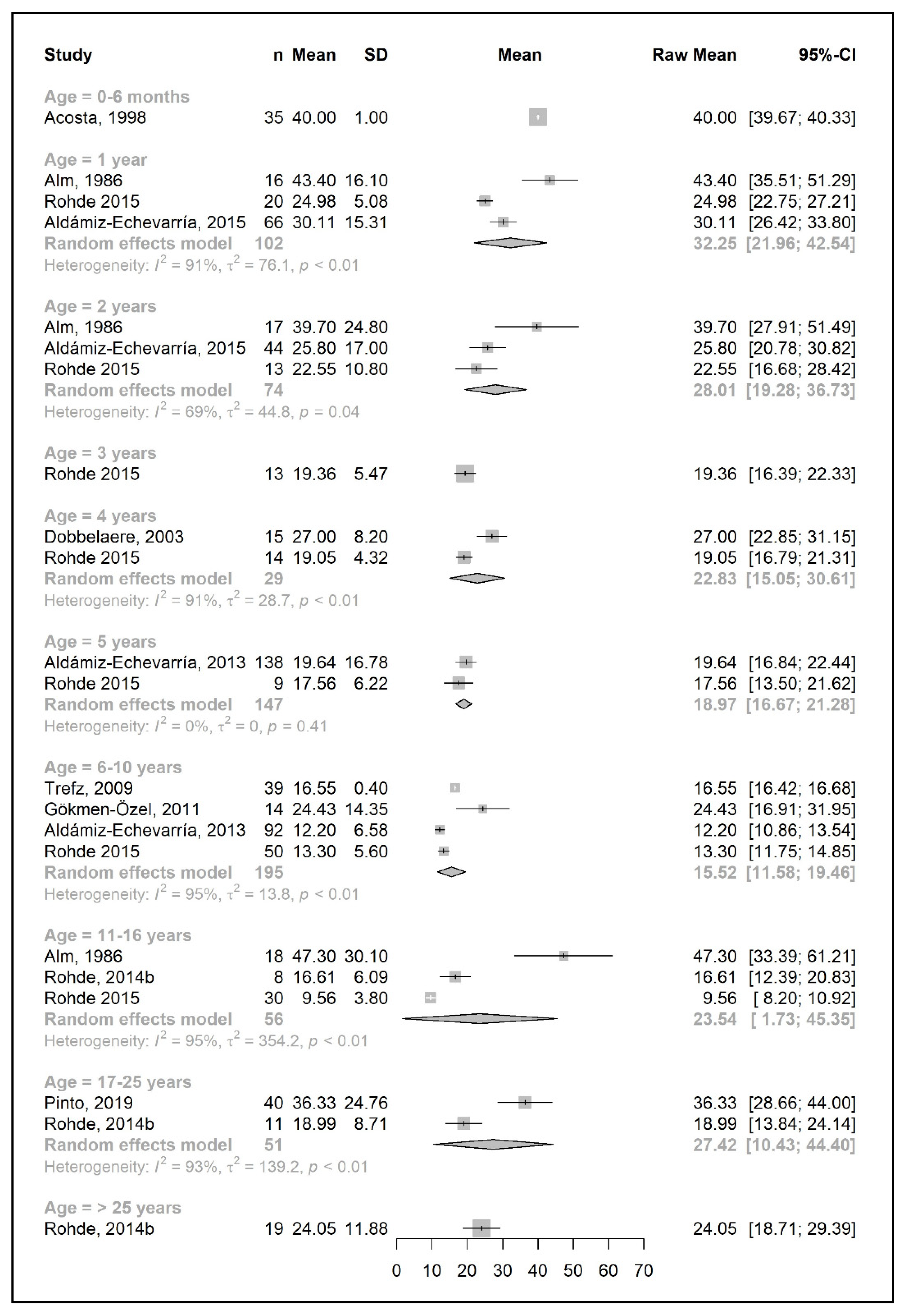
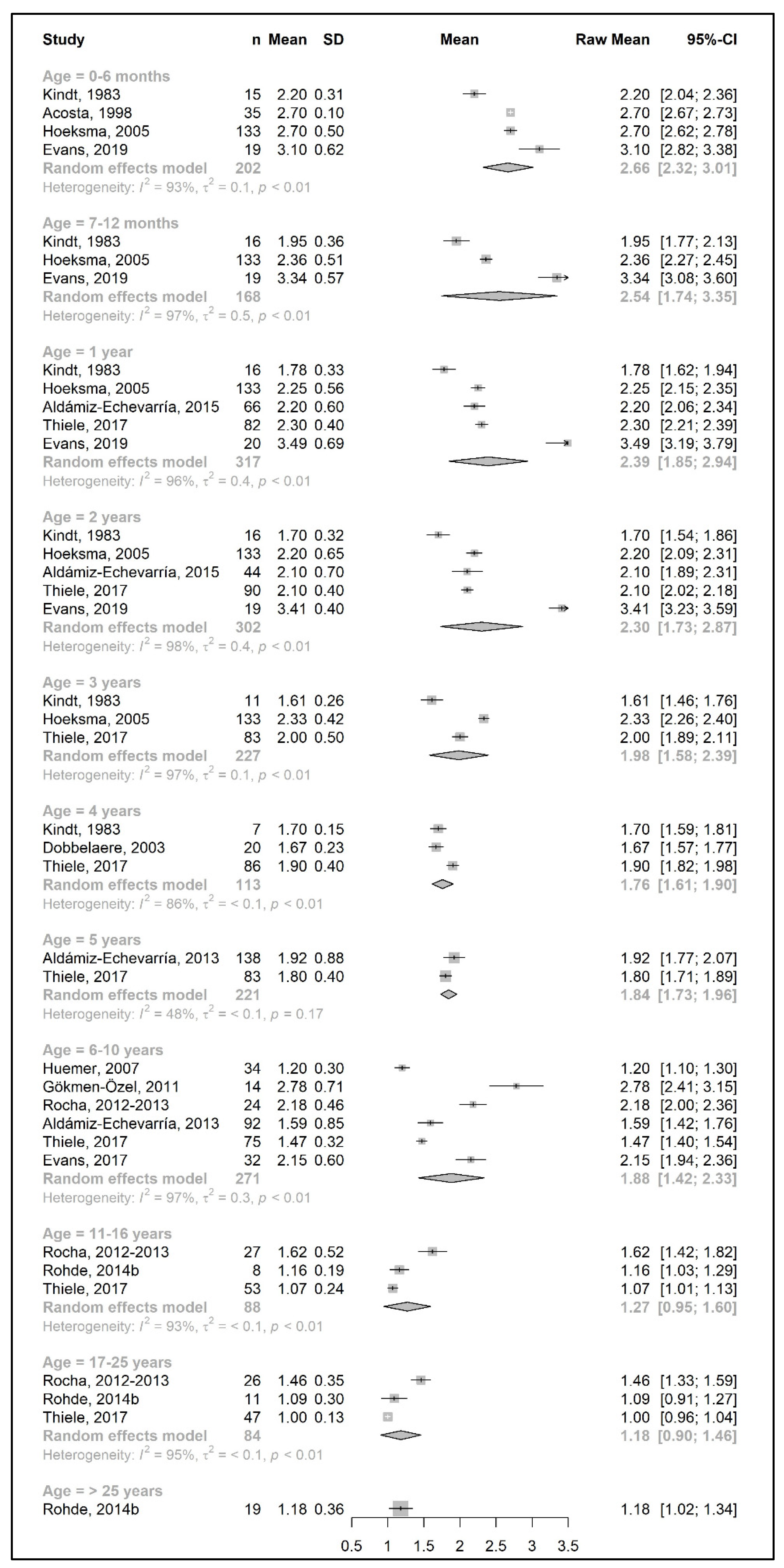
3.3. Primary Outcomes: Changes in Phe, Natural Protein, and Total Protein Intakes throughout Lifetime
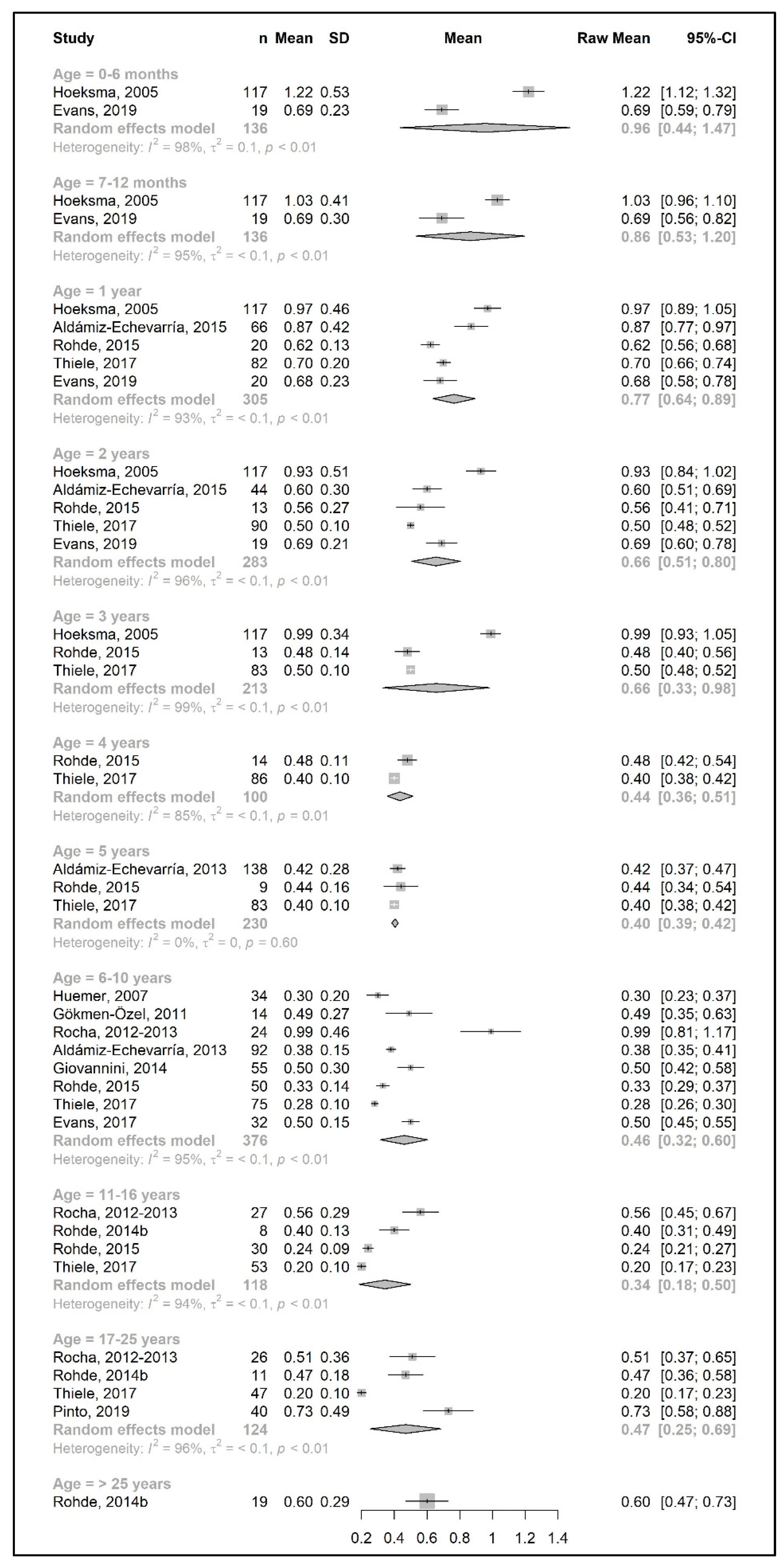
3.3.1. Phenylalanine and Natural Protein Tolerance
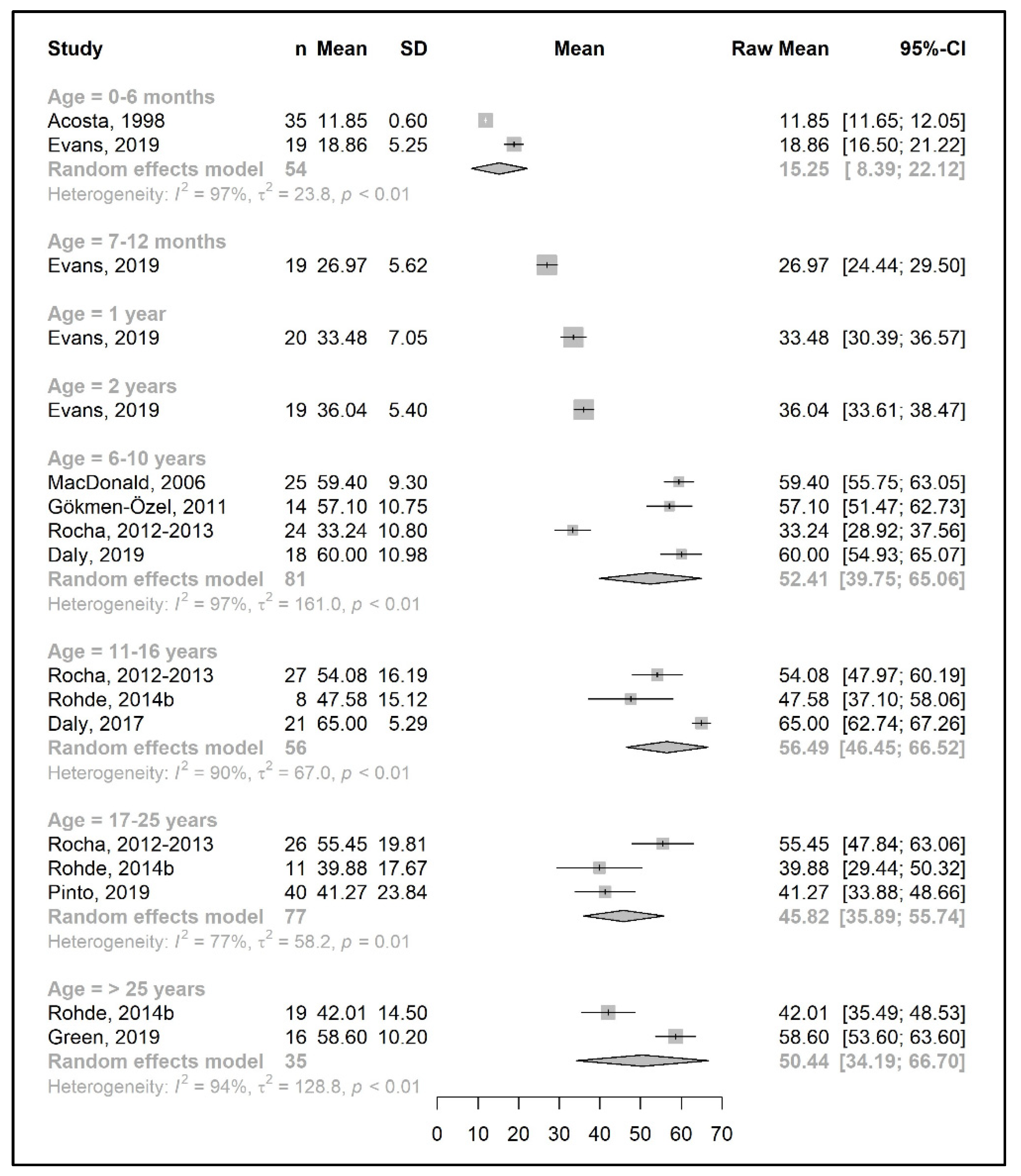
3.3.2. Protein Equivalent from Protein Substitute Intake
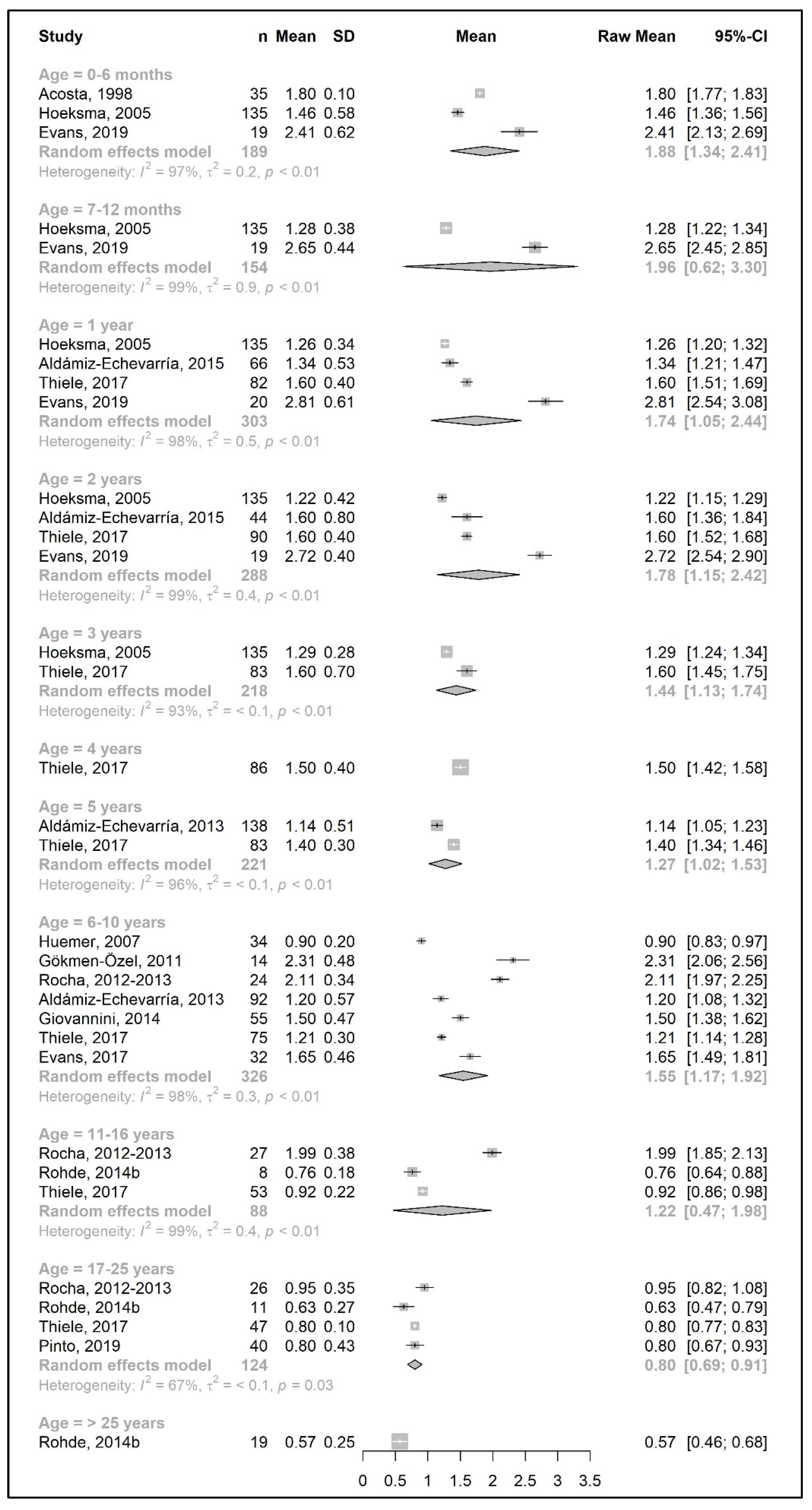
3.3.3. Total Protein Intake
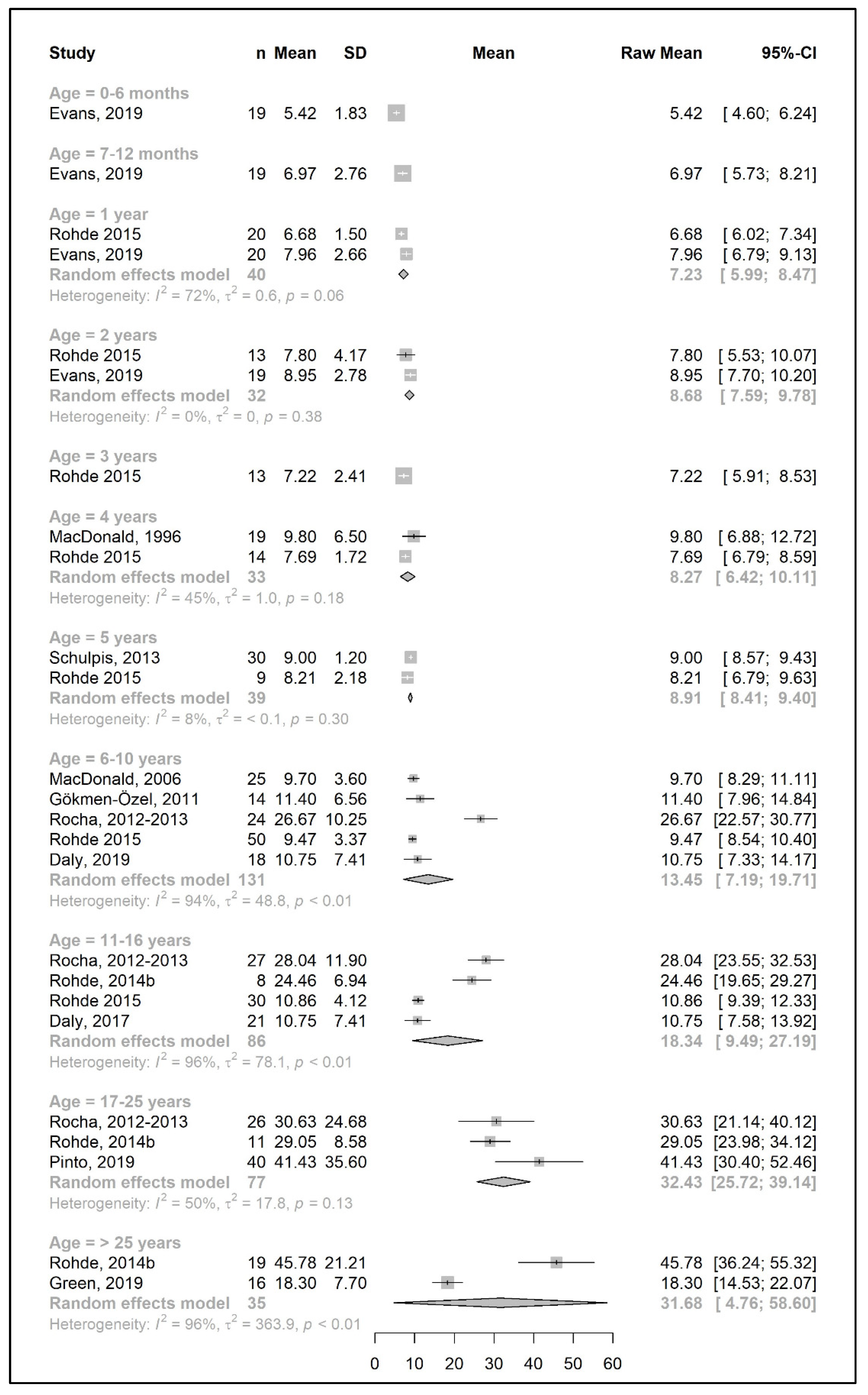
3.4. Secondary Outcomes: Energy and Metabolic Control in Patients with PKU
3.4.1. Energy Intake
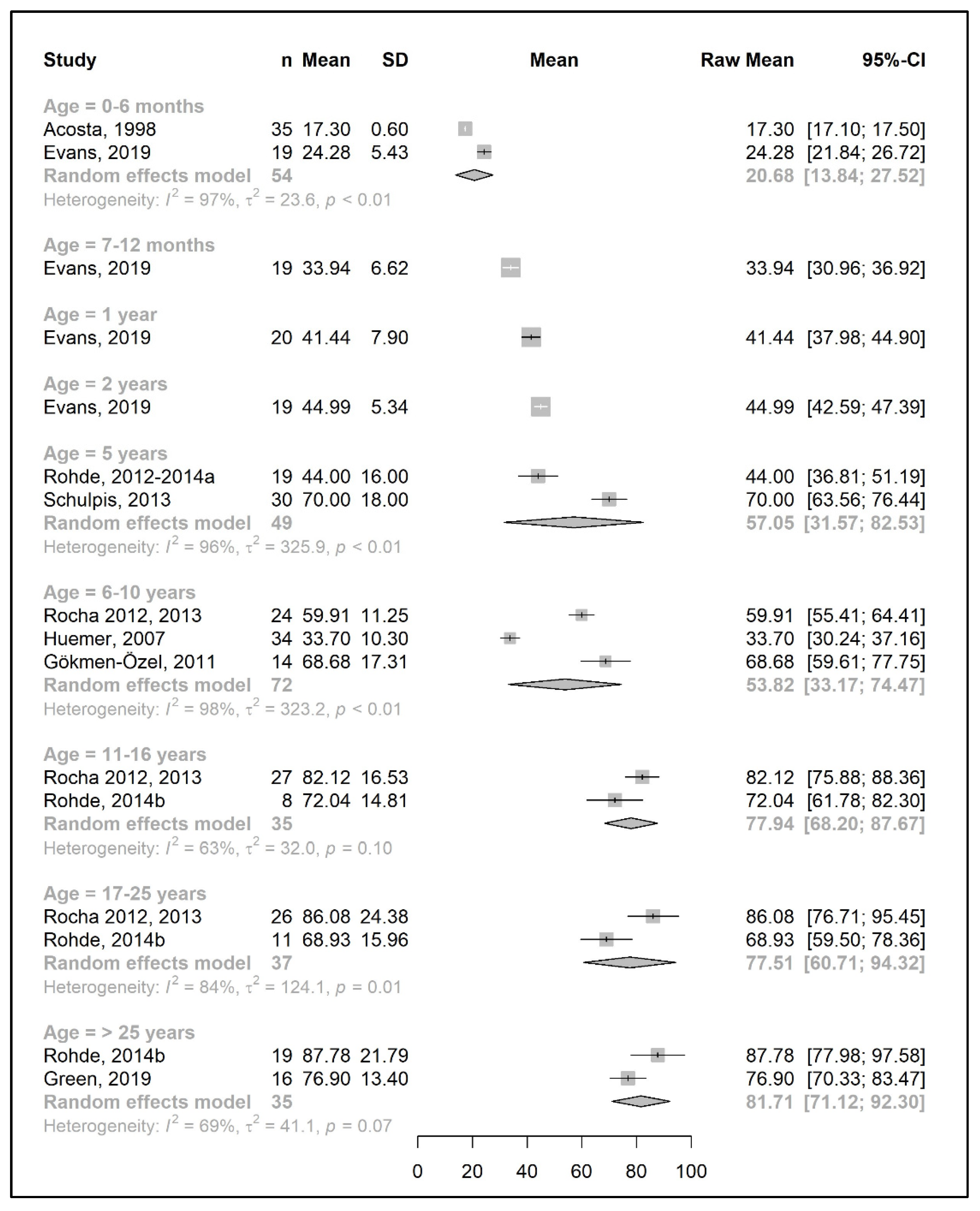
3.4.2. Metabolic Control
3.5. Quality Appraisal and Risk of Bias Assessment
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; van Wegberg, A.M.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Courtney-Martin, G.; Bross, R.; Raffi, M.; Clarke, J.T.; Ball, R.O.; Pencharz, P.B. Phenylalanine requirement in children with classical PKU determined by indicator amino acid oxidation. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E1249–E1256. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; van Wegberg, A.M.J.; Ahring, K.; Beblo, S.; Bélanger-Quintana, A.; Burlina, A.; Campistol, J.; Coşkun, T.; Feillet, F.; Giżewska, M.; et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J. Rare Dis. 2020, 15, 171. [Google Scholar] [CrossRef]
- Burnett, J.R. Sapropterin dihydrochloride (Kuvan/phenoptin), an orally active synthetic form of BH4 for the treatment of phenylketonuria. IDrugs 2007, 10, 805–813. [Google Scholar] [PubMed]
- Hydery, T.; Coppenrath, V.A. A Comprehensive Review of Pegvaliase, an Enzyme Substitution Therapy for the Treatment of Phenylketonuria. Drug Target Insights 2019, 13, 1177392819857089. [Google Scholar] [CrossRef]
- Ilgaz, F.; Marsaux, C.; Pinto, A.; Singh, R.; Rohde, C.; Karabulut, E.; Gökmen-Özel, H.; Kuhn, M.; MacDonald, A. Protein Substitute Requirements of Patients with Phenylketonuria on BH4 Treatment: A Systematic Review and Meta-Analysis. Nutrients 2021, 13, 1040. [Google Scholar] [CrossRef] [PubMed]
- Lindegren, M.L.; Krishnaswami, S.; Reimschisel, T.; Fonnesbeck, C.; Sathe, N.A.; McPheeters, M.L. A Systematic Review of BH4 (Sapropterin) for the Adjuvant Treatment of Phenylketonuria. JIMD Rep. 2013, 8, 109–119. [Google Scholar] [CrossRef]
- Somaraju, U.R.; Merrin, M. Sapropterin dihydrochloride for phenylketonuria. Cochrane Database Syst. Rev. 2015, 2015, Cd008005. [Google Scholar] [CrossRef]
- Burton, B.K.; Longo, N.; Vockley, J.; Grange, D.K.; Harding, C.O.; Decker, C.; Li, M.; Lau, K.; Rosen, O.; Larimore, K.; et al. Pegvaliase for the treatment of phenylketonuria: Results of the phase 2 dose-finding studies with long-term follow-up. Mol. Genet. Metab. 2020, 130, 239–246. [Google Scholar] [CrossRef]
- van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef]
- Fernstrom, J.D.; Fernstrom, M.H. Tyrosine, phenylalanine, and catecholamine synthesis and function in the brain. J. Nutr. 2007, 137, 1539S–1547S, discussion 1548S. [Google Scholar] [CrossRef]
- MacDonald, A.; Rocha, J.C.; van Rijn, M.; Feillet, F. Nutrition in phenylketonuria. Mol. Genet. Metab. 2011, 104, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Hanley, W.B.; Linsao, L.; Davidson, W.; Moes, C.A. Malnutrition with early treatment of phenylketonuria. Pediatr. Res. 1970, 4, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Trefz, F.; Maillot, F.; Motzfeldt, K.; Schwarz, M. Adult phenylketonuria outcome and management. Mol. Genet. Metab. 2011, 104, S26–S30. [Google Scholar] [CrossRef]
- MacDonald, A. Diet and phenylketonuria: Time for change? J. Hum. Nutr. Diet. 1994, 7, 105–114. [Google Scholar] [CrossRef]
- MacDonald, A.; Rylance, G.; Davies, P.; Asplin, D.; Hall, S.K.; Booth, I.W. Free use of fruits and vegetables in phenylketonuria. J. Inherit. Metab. Dis. 2003, 26, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Mütze, U.; Schulz, S.; Thiele, A.G.; Ceglarek, U.; Thiery, J.; Mueller, A.S.; Kiess, W.; Beblo, S. Unrestricted fruits and vegetables in the PKU diet: A 1-year follow-up. Eur. J. Clin. Nutr. 2014, 68, 401–403. [Google Scholar] [CrossRef]
- Rohde, C.; Mütze, U.; Weigel, J.F.; Ceglarek, U.; Thiery, J.; Kiess, W.; Beblo, S. Unrestricted consumption of fruits and vegetables in phenylketonuria: No major impact on metabolic control. Eur. J. Clin. Nutr. 2012, 66, 633–638. [Google Scholar] [CrossRef]
- Zimmermann, M.; Jacobs, P.; Fingerhut, R.; Torresani, T.; Thöny, B.; Blau, N.; Baumgartner, M.R.; Rohrbach, M. Positive effect of a simplified diet on blood phenylalanine control in different phenylketonuria variants, characterized by newborn BH4 loading test and PAH analysis. Mol. Genet. Metab. 2012, 106, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- National Heart, Lung and Blood Institute (NHLBI) Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies. Available online: https://www.nhlbi.nih.gov/health-topics/study-quality-assessment-tools (accessed on 9 May 2023).
- R Development Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Rocha, J.C.; van Spronsen, F.J.; Almeida, M.F.; Ramos, E.; Guimarães, J.T.; Borges, N. Early dietary treated patients with phenylketonuria can achieve normal growth and body composition. Mol. Genet. Metab. 2013, 110, S40–S43. [Google Scholar] [CrossRef]
- Rocha, J.C.; van Spronsen, F.J.; Almeida, M.F.; Soares, G.; Quelhas, D.; Ramos, E.; Guimarães, J.T.; Borges, N. Dietary treatment in phenylketonuria does not lead to increased risk of obesity or metabolic syndrome. Mol. Genet. Metab. 2012, 107, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Alm, J.; Bodegård, G.; Larsson, A.; Nyberg, G.; Zetterström, R. Children with inborn errors of phenylalanine metabolism: Prognosis and phenylalanine tolerance. Acta Paediatr. Scand. 1986, 75, 619–625. [Google Scholar] [CrossRef]
- Evers, R.A.F.; van Wegberg, A.M.J.; van Dam, E.; de Vries, M.C.; Janssen, M.C.H.; van Spronsen, F.J. Anthropomorphic measurements and nutritional biomarkers after 5 years of BH(4) treatment in phenylketonuria patients. Mol. Genet. Metab. 2018, 124, 238–242. [Google Scholar] [CrossRef]
- Pinto, A.; Almeida, M.F.; MacDonald, A.; Ramos, P.C.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; Jackson, R.; et al. Over Restriction of Dietary Protein Allowance: The Importance of Ongoing Reassessment of Natural Protein Tolerance in Phenylketonuria. Nutrients 2019, 11, 995. [Google Scholar] [CrossRef]
- Sweeney, A.L.; Roberts, R.M.; Fletcher, J.M. Dietary protein counting as an alternative way of maintaining metabolic control in phenylketonuria. JIMD Rep. 2012, 3, 131–139. [Google Scholar] [CrossRef]
- Acosta, P.B.; Yannicelli, S.; Marriage, B.; Mantia, C.; Gaffield, B.; Porterfield, M.; Hunt, M.; McMaster, N.; Bernstein, L.; Parton, P.; et al. Nutrient intake and growth of infants with phenylketonuria undergoing therapy. J. Pediatr. Gastroenterol. Nutr. 1998, 27, 287–291. [Google Scholar] [CrossRef]
- Aldámiz-Echevarría, L.; Bueno, M.A.; Couce, M.L.; Lage, S.; Dalmau, J.; Vitoria, I.; Andrade, F.; Blasco, J.; Alcalde, C.; Gil, D.; et al. Anthropometric characteristics and nutrition in a cohort of PAH-deficient patients. Clin. Nutr. 2014, 33, 702–717. [Google Scholar] [CrossRef]
- Aldámiz-Echevarría, L.; Bueno, M.A.; Couce, M.L.; Lage, S.; Dalmau, J.; Vitoria, I.; Andrade, F.; Llarena, M.; Blasco, J.; Alcalde, C.; et al. Tetrahydrobiopterin therapy vs phenylalanine-restricted diet: Impact on growth in PKU. Mol. Genet. Metab. 2013, 109, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Aldámiz-Echevarría, L.; Bueno, M.A.; Couce, M.L.; Lage, S.; Dalmau, J.; Vitoria, I.; Llarena, M.; Andrade, F.; Blasco, J.; Alcalde, C.; et al. 6R-tetrahydrobiopterin treated PKU patients below 4 years of age: Physical outcomes, nutrition and genotype. Mol. Genet. Metab. 2015, 115, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.; Evans, S.; Chahal, S.; Santra, S.; MacDonald, A. Glycomacropeptide in children with phenylketonuria: Does its phenylalanine content affect blood phenylalanine control? J. Hum. Nutr. Diet. 2017, 30, 515–523. [Google Scholar] [CrossRef]
- Daly, A.; Evans, S.; Chahal, S.; Santra, S.; Pinto, A.; Gingell, C.; Rocha, J.C.; van Spronsen, F.; Jackson, R.; MacDonald, A. The Effect of Glycomacropeptide versus Amino Acids on Phenylalanine and Tyrosine Variability over 24 Hours in Children with PKU: A Randomized Controlled Trial. Nutrients 2019, 11, 520. [Google Scholar] [CrossRef]
- Dobbelaere, D.; Michaud, L.; Debrabander, A.; Vanderbecken, S.; Gottrand, F.; Turck, D.; Farriaux, J.P. Evaluation of nutritional status and pathophysiology of growth retardation in patients with phenylketonuria. J. Inherit. Metab. Dis. 2003, 26, 1–11. [Google Scholar] [CrossRef]
- Evans, M.; Truby, H.; Boneh, A. The relationship between dietary intake, growth and body composition in Phenylketonuria. Mol. Genet. Metab. 2017, 122, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.; Daly, A.; Wildgoose, J.; Cochrane, B.; Chahal, S.; Ashmore, C.; Loveridge, N.; MacDonald, A. Growth, Protein and Energy Intake in Children with PKU Taking a Weaning Protein Substitute in the First Two Years of Life: A Case-Control Study. Nutrients 2019, 11, 552. [Google Scholar] [CrossRef]
- Giovannini, M.; Riva, E.; Salvatici, E.; Cefalo, G.; Radaelli, G. Randomized controlled trial of a protein substitute with prolonged release on the protein status of children with phenylketonuria. J. Am. Coll. Nutr. 2014, 33, 103–110. [Google Scholar] [CrossRef]
- Gokmen-Ozel, H.; Ferguson, C.; Evans, S.; Daly, A.; MacDonald, A. Does a lower carbohydrate protein substitute impact on blood phenylalanine control, growth and appetite in children with PKU? Mol. Genet. Metab. 2011, 104, S64–S67. [Google Scholar] [CrossRef]
- Green, B.; Browne, R.; Firman, S.; Hill, M.; Rahman, Y.; Kaalund Hansen, K.; Adam, S.; Skeath, R.; Hallam, P.; Herlihy, I.; et al. Nutritional and Metabolic Characteristics of UK Adult Phenylketonuria Patients with Varying Dietary Adherence. Nutrients 2019, 11, 2459. [Google Scholar] [CrossRef]
- Hoeksma, M.; Van Rijn, M.; Verkerk, P.H.; Bosch, A.M.; Mulder, M.F.; de Klerk, J.B.; de Koning, T.J.; Rubio-Gozalbo, E.; de Vries, M.; Sauer, P.J.; et al. The intake of total protein, natural protein and protein substitute and growth of height and head circumference in Dutch infants with phenylketonuria. J. Inherit. Metab. Dis. 2005, 28, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Huemer, M.; Huemer, C.; Möslinger, D.; Huter, D.; Stöckler-Ipsiroglu, S. Growth and body composition in children with classical phenylketonuria: Results in 34 patients and review of the literature. J. Inherit. Metab. Dis. 2007, 30, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Kindt, E.; Motzfeldt, K.; Halvorsen, S.; Lie, S.O. Protein requirements in infants and children: A longitudinal study of children treated for phenylketonuria. Am. J. Clin. Nutr. 1983, 37, 778–785. [Google Scholar] [CrossRef]
- MacDonald, A.; Chakrapani, A.; Hendriksz, C.; Daly, A.; Davies, P.; Asplin, D.; Hall, K.; Booth, I.W. Protein substitute dosage in PKU: How much do young patients need? Arch. Dis. Child. 2006, 91, 588–593. [Google Scholar] [CrossRef][Green Version]
- Ponzone, A.; Spada, M.; Roasio, L.; Porta, F.; Mussa, A.; Ferraris, S. Impact of neonatal protein metabolism and nutrition on screening for phenylketonuria. J. Pediatr. Gastroenterol. Nutr. 2008, 46, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Thiele, A.G.; Och, U.; Schönherr, K.; Meyer, U.; Rosenbaum-Fabian, S.; Maddalon, C.; Matzken, S.; Blessing, H.; Lang, F.; et al. Effect of dietary regime on metabolic control in phenylketonuria: Is exact calculation of phenylalanine intake really necessary? Mol. Genet. Metab. Rep. 2015, 5, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; von Teeffelen-Heithoff, A.; Thiele, A.G.; Arelin, M.; Mütze, U.; Kiener, C.; Gerloff, J.; Baerwald, C.; Schultz, S.; Heller, C.; et al. PKU patients on a relaxed diet may be at risk for micronutrient deficiencies. Eur. J. Clin. Nutr. 2014, 68, 119–124. [Google Scholar] [CrossRef]
- Schulpis, K.H.; Gioni, V.; Platis, D.; Kalogerakou, M.; Karikas, G.A.; Afordakou, D.; Papassotiriou, I. The beneficial effect of adherence to the therapeutic diet on the main protein-energy wasting and several other biomarkers in children with phenylketonuria. J. Pediatr. Biochem. 2013, 3, 99–106. [Google Scholar] [CrossRef]
- Thiele, A.G.; Gausche, R.; Lindenberg, C.; Beger, C.; Arelin, M.; Rohde, C.; Mütze, U.; Weigel, J.F.; Mohnike, K.; Baerwald, C.; et al. Growth and Final Height Among Children With Phenylketonuria. Pediatrics 2017, 140, e20170015. [Google Scholar] [CrossRef]
- Trefz, F.K.; Burton, B.K.; Longo, N.; Casanova, M.M.; Gruskin, D.J.; Dorenbaum, A.; Kakkis, E.D.; Crombez, E.A.; Grange, D.K.; Harmatz, P.; et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: A phase III, randomized, double-blind, placebo-controlled study. J. Pediatr. 2009, 154, 700–707. [Google Scholar] [CrossRef]
- Evans, S.; Daly, A.; MacDonald, J.; Pinto, A.; MacDonald, A. Fifteen years of using a second stage protein substitute for weaning in phenylketonuria: A retrospective study. J. Hum. Nutr. Diet. 2018, 31, 349–356. [Google Scholar] [CrossRef]
- Ferguson, C. Monitoring the effect of varying the distribution of phenylalanine exchanges and protein substitute on serum phenylalanine—A preliminary study. J. Hum. Nutr. Diet. 1996, 9, 293–302. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; van Rijn, M.; Dorgelo, B.; Hoeksma, M.; Bosch, A.M.; Mulder, M.F.; de Klerk, J.B.; de Koning, T.; Rubio-Gozalbo, M.E.; de Vries, M.; et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. J. Inherit. Metab. Dis. 2009, 32, 27–31. [Google Scholar] [CrossRef]
- Wendel, U.; Ullrich, K.; Schmidt, H.; Batzler, U. Six-year follow up of phenylalanine intakes and plasma phenylalanine concentrations. Eur. J. Pediatr. 1990, 149 (Suppl. S1), S13–S16. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; Rylance, G.; Hall, S.K.; Asplin, D.; Booth, I.W. Factors affecting the variation in plasma phenylalanine in patients with phenylketonuria on diet. Arch. Dis. Child. 1996, 74, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Stockler-Ipsiroglu, S.; Yuskiv, N.; Salvarinova, R.; Apatean, D.; Ho, G.; Cheng, B.; Giezen, A.; Lillquist, Y.; Ueda, K. Individualized long-term outcomes in blood phenylalanine concentrations and dietary phenylalanine tolerance in 11 patients with primary phenylalanine hydroxylase (PAH) deficiency treated with Sapropterin-dihydrochloride. Mol. Genet. Metab. 2015, 114, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Food and Nutrition Board of the Institute of Medicine, The National Academies. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholestetrol, Protein and Amino Acids; The National Academies Press: Washington, DC, USA, 2005. [Google Scholar]
- World Health Organization. Protein and Amino Acid Requirements in Human Nutrition; World Health Organization Technical Report Series; World Health Organization: Geneva, Switzerland, 2007; pp. 1–265.
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef]
- MacLeod, E.L.; Gleason, S.T.; van Calcar, S.C.; Ney, D.M. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Mol. Genet. Metab. 2009, 98, 331–337. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, M.; Hoeksma, M.; Sauer, P.J.; Modderman, P.; Reijngoud, D.J.; van Spronsen, F.J. Adult patients with well-controlled phenylketonuria tolerate incidental additional intake of phenylalanine. Ann. Nutr. Metab. 2011, 58, 94–100. [Google Scholar] [CrossRef]
- Macdonald, A.; Davies, P.; Daly, A.; Hopkins, V.; Hall, S.K.; Asplin, D.; Hendriksz, C.; Chakrapani, A. Does maternal knowledge and parent education affect blood phenylalanine control in phenylketonuria? J. Hum. Nutr. Diet. 2008, 21, 351–358. [Google Scholar] [CrossRef]
- van Rijn, M.; Jansma, J.; Brinksma, A.; Bakker, H.D.; Boers, G.H.; Carbasius-Weber, E.; Douwes, A.C.; van den Herberg, A.; Ter Horst, N.M.; de Klerk, J.B.; et al. A survey of natural protein intake in Dutch phenylketonuria patients: Insight into estimation or measurement of dietary intake. J. Am. Diet. Assoc. 2008, 108, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.; Pinto, A.; Evans, S.; Daly, A.; Ashmore, C.; Ford, S.; Buckley, S.; MacDonald, A. The Challenges and Dilemmas of Interpreting Protein Labelling of Prepackaged Foods Encountered by the PKU Community. Nutrients 2022, 14, 1355. [Google Scholar] [CrossRef]
- Snyderman, S.E.; Holt, L.E.; Norton, P.M.; Roitman, E.; Phansalkar, S.V. The Plasma Aminogram. I. Influence of the Level of Protein Intake and a Comparison of Whole Protein and Amino Acid Diets. Pediatr. Res. 1968, 2, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.; Daly, A.; Chahal, S.; MacDonald, J.; MacDonald, A. Food acceptance and neophobia in children with phenylketonuria: A prospective controlled study. J. Hum. Nutr. Diet. 2016, 29, 427–433. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; Harris, G.; Rylance, G.; Asplin, D.; Booth, I.W. Abnormal feeding behaviours in phenylketonuria. J. Hum. Nutr. Diet. 1997, 10, 163–170. [Google Scholar] [CrossRef]
- Garza, C.; Scrimshaw, N.S.; Young, V.R. Human protein requirements: The effect of variations in energy intake within the maintenance range. Am. J. Clin. Nutr. 1976, 29, 280–287. [Google Scholar] [CrossRef]
- Zello, G.A.; Pencharz, P.B.; Ball, R.O. Phenylalanine flux, oxidation, and conversion to tyrosine in humans studied with L-[1-13C]phenylalanine. Am. J. Physiol.-Endocrinol. Metab. 1990, 259, E835–E843. [Google Scholar] [CrossRef]
- Dangin, M.; Guillet, C.; Garcia-Rodenas, C.; Gachon, P.; Bouteloup-Demange, C.; Reiffers-Magnani, K.; Fauquant, J.; Ballèvre, O.; Beaufrère, B. The rate of protein digestion affects protein gain differently during aging in humans. J. Physiol. 2003, 549, 635–644. [Google Scholar] [CrossRef]
- Reginster, J.Y.; Beaudart, C.; Al-Daghri, N.; Avouac, B.; Bauer, J.; Bere, N.; Bruyère, O.; Cerreta, F.; Cesari, M.; Rosa, M.M.; et al. Update on the ESCEO recommendation for the conduct of clinical trials for drugs aiming at the treatment of sarcopenia in older adults. Aging Clin. Exp. Res. 2021, 33, 3–17. [Google Scholar] [CrossRef]
- Firman, S.; Witard, O.C.; O’Keeffe, M.; Ramachandran, R. Dietary protein and protein substitute requirements in adults with phenylketonuria: A review of the clinical guidelines. Clin. Nutr. 2021, 40, 702–709. [Google Scholar] [CrossRef]
- Aguiar, A.; Ahring, K.; Almeida, M.F.; Assoun, M.; Belanger Quintana, A.; Bigot, S.; Bihet, G.; Blom Malmberg, K.; Burlina, A.; Bushueva, T.; et al. Practices in prescribing protein substitutes for PKU in Europe: No uniformity of approach. Mol. Genet. Metab. 2015, 115, 17–22. [Google Scholar] [CrossRef]
- Ford, S.; O’Driscoll, M.; MacDonald, A. Living with Phenylketonuria: Lessons from the PKU community. Mol. Genet. Metab. Rep. 2018, 17, 57–63. [Google Scholar] [CrossRef]
- Ilgaz, F.; Pinto, A.; Gökmen-Özel, H.; Rocha, J.C.; van Dam, E.; Ahring, K.; Bélanger-Quintana, A.; Dokoupil, K.; Karabulut, E.; MacDonald, A. Long-Term Growth in Phenylketonuria: A Systematic Review and Meta-Analysis. Nutrients 2019, 11, 2070. [Google Scholar] [CrossRef] [PubMed]
- Duran, G.P.; Rohr, F.J.; Slonim, A.; Güttler, F.; Levy, H.L. Necessity of complete intake of phenylalanine-free amino acid mixture for metabolic control of phenylketonuria. J. Am. Diet. Assoc. 1999, 99, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Scheinin, M.; Barassi, A.; Junnila, J.; Lovró, Z.; Reiner, G.; Sarkkinen, E.; MacDonald, A. Amino Acid Plasma Profiles from a Prolonged-Release Protein Substitute for Phenylketonuria: A Randomized, Single-Dose, Four-Way Crossover Trial in Healthy Volunteers. Nutrients 2020, 12, 1653. [Google Scholar] [CrossRef] [PubMed]
- Acosta, P.B.; Yannicelli, S. Protein intake affects phenylalanine requirements and growth of infants with phenylketonuria. Acta Paediatr. 1994, 407, 66–67. [Google Scholar] [CrossRef]
- Grünert, S.C.; Brichta, C.M.; Krebs, A.; Clement, H.W.; Rauh, R.; Fleischhaker, C.; Hennighausen, K.; Sass, J.O.; Schwab, K.O. Diurnal variation of phenylalanine and tyrosine concentrations in adult patients with phenylketonuria: Subcutaneous microdialysis is no adequate tool for the determination of amino acid concentrations. Nutr. J. 2013, 12, 60. [Google Scholar] [CrossRef]
- Mazzola, P.N.; Teixeira, B.C.; Schirmbeck, G.H.; Reischak-Oliveira, A.; Derks, T.G.J.; van Spronsen, F.J.; Dutra-Filho, C.S.; Schwartz, I.V.D. Acute exercise in treated phenylketonuria patients: Physical activity and biochemical response. Mol. Genet. Metab. Rep. 2015, 5, 55–59. [Google Scholar] [CrossRef]
- Pugliese, M.; Tingley, K.; Chow, A.; Pallone, N.; Smith, M.; Chakraborty, P.; Geraghty, M.T.; Irwin, J.K.; Mitchell, J.J.; Stockler, S.; et al. Core Outcome Sets for Medium-Chain Acyl-CoA Dehydrogenase Deficiency and Phenylketonuria. Pediatrics 2021, 148, e2020037747. [Google Scholar] [CrossRef]
- Pinto, A.; Adams, S.; Ahring, K.; Allen, H.; Almeida, M.F.; Garcia-Arenas, D.; Arslan, N.; Assoun, M.; Atik Altınok, Y.; Barrio-Carreras, D.; et al. Weaning practices in phenylketonuria vary between health professionals in Europe. Mol. Genet. Metab. Rep. 2019, 18, 39–44. [Google Scholar] [CrossRef]









| NIH Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies | Research Question | Study Population | Participant Rate at Least 50% from Eligible | Participants from Same/Similar Population | Sample Size Justification | Exposure(s) Measured Prior to the Outcome(s) | Sufficient Time Frame | Examination of Different Levels of Exposure as Related to the Outcome | Definition and Validation of the Exposure Measures | Exposure(s) Assessed More than Once | Definition of Outcome Measures | Blinded Assessors | Loss to Follow-Up after Baseline 20% or Less | Statistical Measure and Adjustment of Key Confounding Variables | Overall Rating |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acosta et al., 1998 [31] | Y | N | ? | Y | N | Y | Y | Y | ? | Y | Y | N | Y | Y | Good |
| Aldámiz-Echevarría et al., 2013 [33] | Y | N | ? | Y | N | Y | Y | Y | Y | Y | Y | N | ? | N | Fair |
| Aldámiz-Echevarría et al., 2014 [32] | Y | Y | ? | Y | N | Y | Y | N | Y | Y | Y | N | ? | N | Fair |
| Aldámiz-Echevarría et al., 2015 [34] | Y | N | ? | Y | N | Y | Y | N | Y | Y | Y | N | Y | N | Fair |
| Alm et al., 1986 [27] | Y | Y | ? | N | N | Y | Y | NA | ? | Y | Y | N | Y | N | Poor |
| Dobbelaere et al., 2003 [37] | Y | N | ? | Y | N | N | N | N | Y | N | Y | N | NA | N | Poor |
| Evans et al., 2017 [38] | Y | Y | ? | Y | N | Y | Y | N | Y | Y | Y | N | NA | N | Fair |
| Evans et al., 2018 [53] | Y | Y | ? | Y | N | Y | Y | Y | Y | Y | Y | N | Y | N | Good |
| Evans et al., 2019 [39] | Y | N | ? | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Fair |
| Evers et al., 2018 [28] | Y | Y | ? | Y | N | Y | Y | N | Y | Y | Y | N | Y | N | Fair |
| Green et al., 2019 [42] | Y | N | ? | Y | N | N | N | Y | Y | N | Y | N | NA | N | Poor |
| Hoeksma et al., 2005 [43] | Y | Y | ? | Y | N | Y | Y | N | Y | Y | Y | N | Y | Y | Good |
| Huemer et al., 2007 [44] | Y | N | ? | Y | N | Y | Y | N | Y | Y | Y | N | Y | N | Fair |
| Kindt et al., 1983 [45] | Y | Y | Y | Y | N | Y | Y | Y | N | Y | N | N | Y | N | Poor |
| MacDonald et al., 1996 [57] | Y | N | ? | ? | N | Y | Y | Y | N | Y | Y | N | Y | N | Fair |
| Pinto et al., 2019 [29] | Y | N | ? | Y | N | Y | Y | N | Y | Y | Y | N | Y | N | Fair |
| Ponzone et al., 2008 [47] | Y | N | ? | Y | N | Y | Y | Y | Y | Y | Y | N | Y | Y | Fair |
| Rocha et al., 2012 & 2013 [25,26] | Y | Y | ? | Y | N | N | N | N | Y | N | Y | N | NA | N | Poor |
| Rohde et al., 2014b [49] | Y | N | ? | N | N | Y | NA | Y | N | N | Y | N | NA | N | Poor |
| Rohde et al., 2015 [48] | Y | N | ? | Y | N | N | N | Y | Y | N | Y | N | ? | N | Poor |
| Schulpis et al., 2013 [50] | Y | N | ? | Y | N | Y | N | Y | Y | Y | Y | N | NA | Y | Fair |
| Stockler-Ipsiroglu [58] | Y | N | ? | Y | N | Y | Y | Y | Y | Y | Y | N | Y | N | Fair |
| Thiele et al., 2017 [51] | Y | Y | Y | Y | N | Y | Y | N | N | Y | Y | N | N | Y | Fair |
| van Spronsen et al., 2009 [55] | Y | Y | Y | Y | N | Y | Y | N | Y | Y | Y | N | Y | N | Fair |
| Wendel et al., 1990 [56] | Y | Y | Y | Y | N | Y | Y | N | Y | Y | N | N | N | N | Poor |
| NIH Quality Assessment of Controlled Intervention Studies | Study Described as a RCT | Adequate Randomization Methods | Concealed Treatment Allocation | Blinded Participants | Blinded Assessors | No Group Differences at Baseline | Low Attrition/Loss of Participants | No Group Differences in Attrition | High Adherence Rates | Avoidance of Interventions Other than the Intended Treatment | Clear, Valid and Reliable Outcome Measures | Sample Size Justification | Prespecified Outcomes and Subgroup Analysis | Use of an Intention-to-Treat Analysis | Overall Ranking |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Daly et al., 2019 [36] | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Good |
| Giovaninni et al., 2014 [40] | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Good |
| Daly et al., 2017 [35] | N | NA | N | N | N | Y | Y | Y | ? | Y | Y | N | Y | Y | Poor |
| Gökmen-Özel et al., 2011 [41] | Y | Y | Y | N | N | Y | ? | ? | ? | Y | Y | Y | Y | Y | Fair |
| MacDonald et al., 2006 [46] | Y | Y | Y | N | N | Y | Y | Y | ? | Y | Y | Y | Y | Y | Good |
| MacDonald et al., 2003 [18] | Y | Y | Y | N | N | Y | Y | Y | ? | Y | N | N | Y | Y | Fair |
| Rohde et al., 2012 & 2014a [19,20] | Y | ? | ? | N | N | Y | Y | Y | ? | Y | Y | N | Y | Y | Fair |
| Trefz et al., 2009 [52] | Y | Y | Y | Y | Y | Y | Y | Y | ? | Y | Y | Y | Y | Y | Good |
| Sweeney et al., 2012 [30] | Y | Y | Y | N | N | Y | Y | Y | ? | Y | Y | ? | Y | Y | Good |
| Ferguson et al., 1996 [54] | N | NA | N | N | N | N | N | N | Y | Y | Y | ? | Y | Y | Poor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Ilgaz, F.; Evans, S.; van Dam, E.; Rocha, J.C.; Karabulut, E.; Hickson, M.; Daly, A.; MacDonald, A. Phenylalanine Tolerance over Time in Phenylketonuria: A Systematic Review and Meta-Analysis. Nutrients 2023, 15, 3506. https://doi.org/10.3390/nu15163506
Pinto A, Ilgaz F, Evans S, van Dam E, Rocha JC, Karabulut E, Hickson M, Daly A, MacDonald A. Phenylalanine Tolerance over Time in Phenylketonuria: A Systematic Review and Meta-Analysis. Nutrients. 2023; 15(16):3506. https://doi.org/10.3390/nu15163506
Chicago/Turabian StylePinto, Alex, Fatma Ilgaz, Sharon Evans, Esther van Dam, Júlio César Rocha, Erdem Karabulut, Mary Hickson, Anne Daly, and Anita MacDonald. 2023. "Phenylalanine Tolerance over Time in Phenylketonuria: A Systematic Review and Meta-Analysis" Nutrients 15, no. 16: 3506. https://doi.org/10.3390/nu15163506
APA StylePinto, A., Ilgaz, F., Evans, S., van Dam, E., Rocha, J. C., Karabulut, E., Hickson, M., Daly, A., & MacDonald, A. (2023). Phenylalanine Tolerance over Time in Phenylketonuria: A Systematic Review and Meta-Analysis. Nutrients, 15(16), 3506. https://doi.org/10.3390/nu15163506