
Potassium Metabolism and Management in Patients with CKD

Abstract
1. Introduction
2. Distribution of K in the Body and Its Roles
3. Regulatory Mechanisms of K in Kidneys
3.1. CCD Intraluminal Flow Velocity and Na Arrival Volume
3.2. ROMK and Maxi-K
3.3. Aldosterone and Kallikrein
3.4. Vasopressin (Arginine Vasopressin: AVP), Insulin, and Glucocorticoids
4. K Transportation in Intestinal Tract
5. Metabolic Regulators of Intracellular K
5.1. Insulin
5.2. Catecholamine
5.3. Intravascular pH
5.4. Osmotic Pressure
6. Epidemiological Results Showing Serum K Levels in Patients with CKD
7. Compensatory Mechanism of K Excretion in Renal Failure Patients
8. Recommended Daily Intake of K
9. Precautions for K Restriction in Elderly Patients with Renal Failure
10. K Restriction Target Value
10.1. Target Value for Patients with Conservative Renal Failure
10.2. Target Value for Hemodialysis Patients
10.3. Target Value for Continuous Hemodialysis Patients
10.4. Target Value for Peritoneal Dialysis Patients
11. Vegetables with Low K Content
12. Evaluation of K Kinetics Using Urinary K Measurement
12.1. Fractional Excretion (FE)
12.2. Transtubular K Gradient (TTKG)
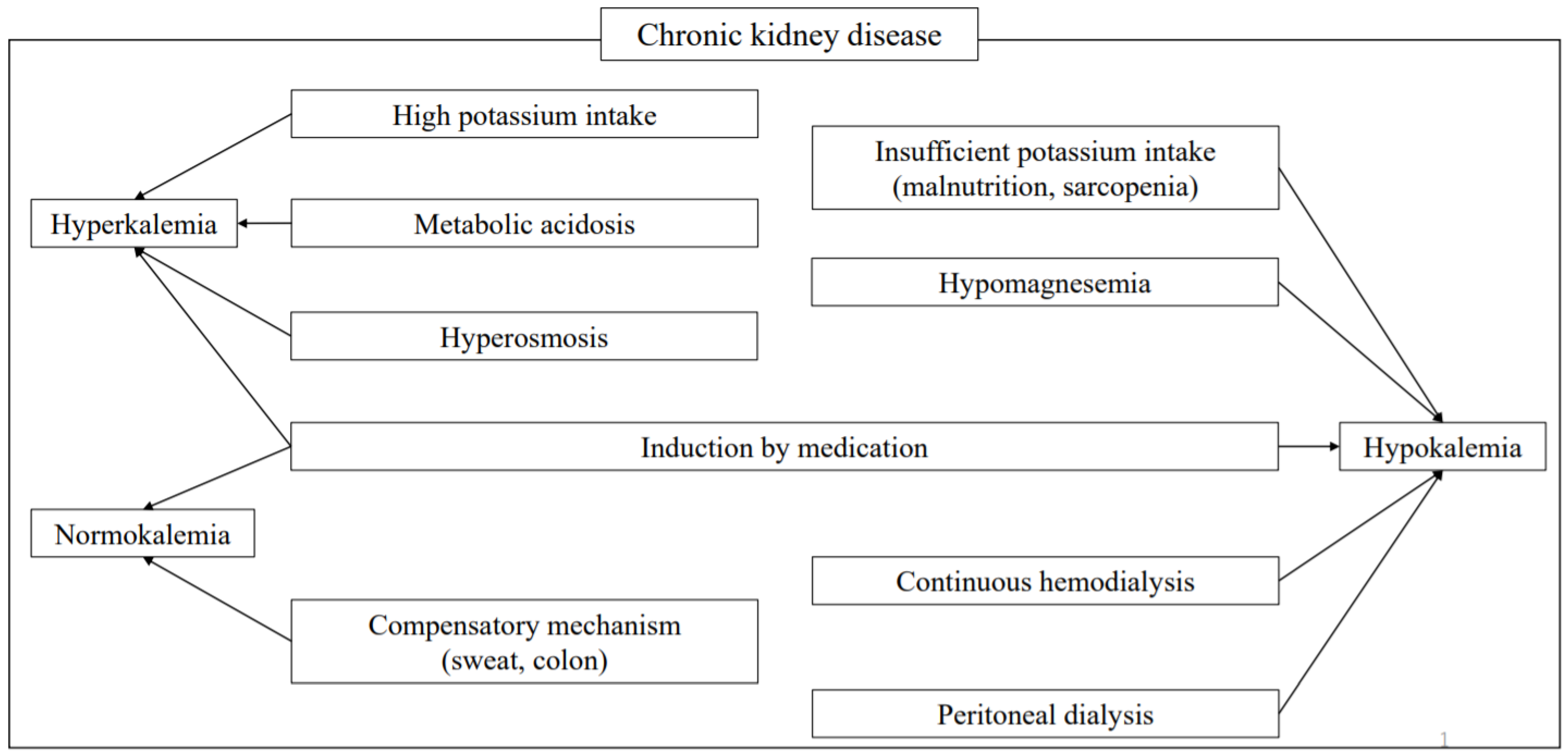
13. Hyperkalemia
13.1. Causes
13.2. Symptoms
13.3. Treatment
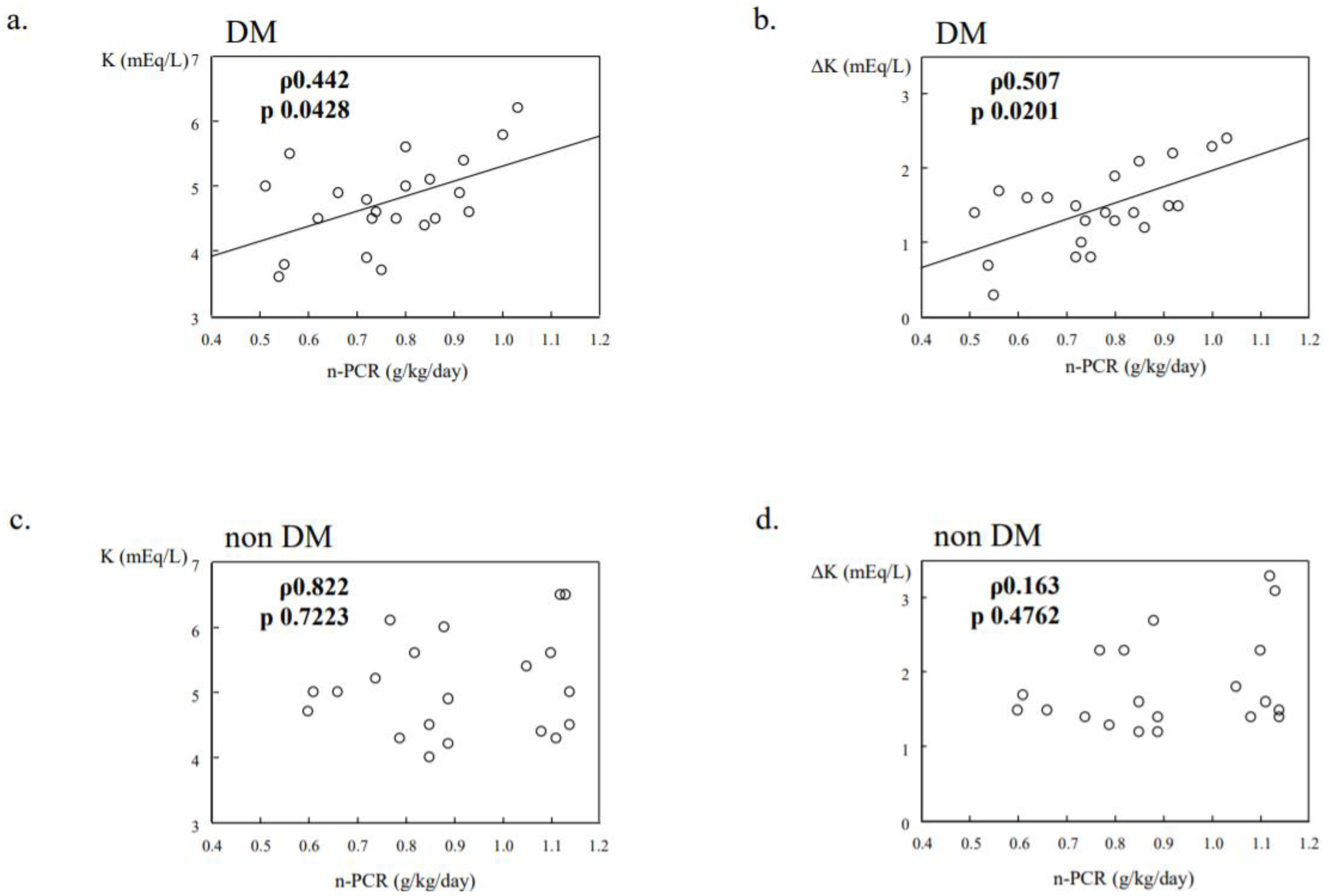
14. K Dynamics in Diabetic Dialysis Patients
15. Hypokalemia
15.1. Causes
15.2. Symptoms
15.3. Treatment
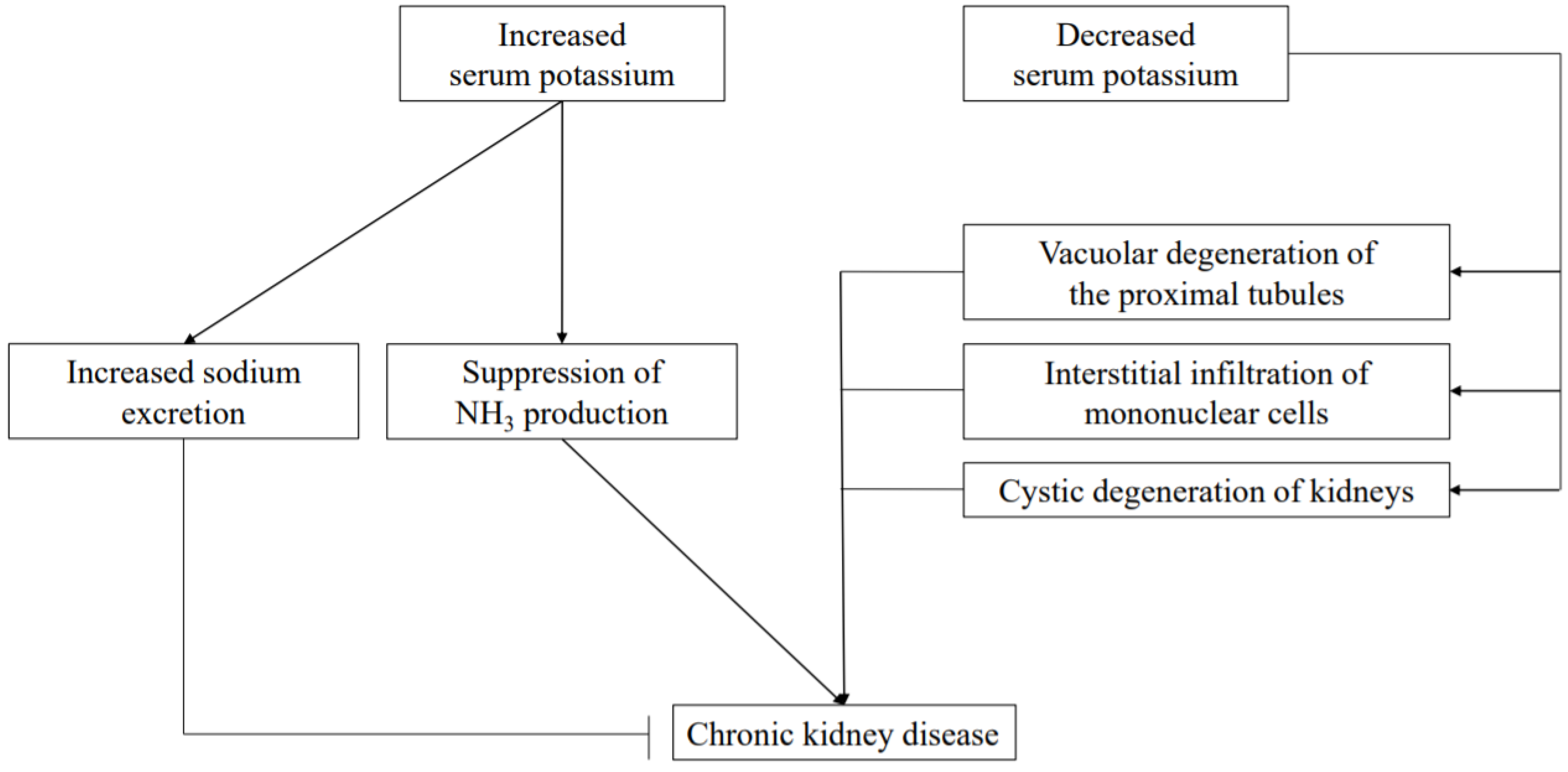
16. Relationship between Mg and K
17. Relationship between NH3 and K
18. Relationship between Na and K
19. Drug-Induced K Abnormalities
19.1. Hyperkalemia
19.2. Hypokalemia
20. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gilligan, S.; Raphael, K.L. Hyperkalemia and Hypokalemia in CKD: Prevalence, Risk Factors, and Clinical Outcomes. Adv. Chronic Kidney Dis. 2017, 24, 315–318. [Google Scholar] [CrossRef]
- Seldin, D.W.; Giebisch, G. (Eds.) The Regulation of Potassium Balance; Raven Press: New York, NY, USA, 1989; pp. 3–29. [Google Scholar]
- Johnson, L.R. (Ed.) Renal regulation of potassium, calcium and magnesium. In Essential Medical Physiology, 3rd ed.; Elsevier Academic Press: San Diego, CA, USA, 2003; pp. 437–446. [Google Scholar]
- Giebisch, G.; Wang, W. Potassium transport: From clearance to channels and pumps. Kidney Int. 1996, 49, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Vallon, V.; Sausbier, M.; Kaissling, B.; Ruth, P.; Osswald, H. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 2007, 72, 566–573. [Google Scholar] [CrossRef]
- Liu, W.; Xu, S.; Woda, C.; Kim, P.; Weinbaum, S.; Satlin, L.M. Effect of flow and stretch on the [Ca2+] response of principle and intercalated cells in cortical collecting duct. Am. J. Physiol. Ren. Physiol. 2003, 285, 998–1012. [Google Scholar] [CrossRef]
- Taniguchi, J.; Tsuruoka, S.; Mizuno, A.; Sato, J.-I.; Fujimura, A.; Suzuki, M. TRPV4 as a flow sensor in flow-dependent K+ secretion from the cortical collecting duct. Am. J. Physiol. Physiol. 2007, 292, F667–F673. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Quigley, R.; Babich, V.; Huang, C.-L. Dietary potassium restriction stimulates endocytosis of ROMK channel in rat cortical collecting duct. Am. J. Physiol. Physiol. 2003, 285, F1179–F1187. [Google Scholar] [CrossRef]
- Wang, W.-H. Regulation of ROMK (Kir1.1) channels: New mechanisms and aspects. Am. J. Physiol. Physiol. 2006, 290, F14–F19. [Google Scholar] [CrossRef] [PubMed]
- Najjar, F.; Zhou, H.; Morimoto, T.; Bruns, J.B.; Li, H.-S.; Liu, W.; Kleyman, T.R.; Satlin, L.M. Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am. J. Physiol. Physiol. 2005, 289, F922–F932. [Google Scholar] [CrossRef]
- Bailey, M.A.; Cantone, A.; Yan, Q.; MacGregor, G.G.; Leng, Q.; Amorim, J.B.O.; Wang, T.; Hebert, S.C.; Giebisch, G.; Malnic, G. Maxi-K channels contribute to urinary potassium excretion in the ROMK deficient mouse model of type H bartter’s syndrome and in adaptation to a high-K diet. Kidney Int. 2006, 70, 51–59. [Google Scholar] [CrossRef]
- Rodan, A.R.; Cheng, C.-J.; Huang, C.-L. Recent advances in distal tubular potassium handling. Am. J. Physiol. Physiol. 2011, 300, F821–F827. [Google Scholar] [CrossRef] [PubMed]
- Giebisch, G.H. A trail of research on potassium. Kidney Int. 2002, 62, 1498–1512. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.N.; Oh, G.; McDonough, A.A.; Youn, J.H. Evidence for gut factor in K+ homeostasis. Am. J. Physiol. Physiol. 2007, 293, F541–F547. [Google Scholar] [CrossRef] [PubMed]
- Va Dinter, T.G.; Fuerst, F.C.; Richardson, C.T.; Ana, C.A.S.; Polter, D.E.; Fordtran, J.S.; Binder, H.J. Stimulated active potassium secretion in a patient with colonic pseudo-obstruction: A new mechanism of secretory diarrhea. Gastroenterology 2005, 129, 1268–1273. [Google Scholar] [CrossRef]
- Sausbier, M.; Matos, J.E.; Sausbier, U.; Beranek, G.; Arntz, C.; Neuhuber, W.; Ruth, P.; Leipziger, J. Distal Colonic K+ Secretion Occurs via BK Channels. J. Am. Soc. Nephrol. 2006, 17, 1275–1282. [Google Scholar] [CrossRef]
- Mathialahan, T.; A MacLennan, K.; Sandle, L.N.; Verbeke, C.; Sandle, G.I. Enhanced large intestinal potassium permeability in end-stage renal disease. J. Pathol. 2005, 206, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Sebata, K.; Watanabe, H.; Shoji, F.; Yamamoto, Y.; Ohashi, M.; Yamada, T.; Matsumoto, H.; Mukouyama, T.; Yonekura, T.; et al. Effect of Oral Glucose Administration on Serum Potassium Concentration in Hemodialysis Patients. Am. J. Kidney Dis. 2005, 46, 697–705. [Google Scholar] [CrossRef]
- Sowinski, K.M.; Cronin, D.; Mueller, B.A.; Kraus, M.A. Subcutaneous terbutamine use in CKD to reduce potassium concentrations. Am. J. Kidney Dis. 2005, 45, 1040–1045. [Google Scholar] [CrossRef]
- Adrogue, H.J.; Masias, N.E. Change in plasma potassium concentration during acute acid-base disturbances. Am. J. Med. 1981, 71, 456–467. [Google Scholar] [CrossRef]
- Kashihara, N.; Kohsaka, S.; Kanda, E.; Okami, S.; Yajima, T. Hyperkalemia in Real-World Patients Under Continuous Medical Care in Japan. Kidney Int. Rep. 2019, 4, 1248–1260. [Google Scholar] [CrossRef]
- Sofue, T.; Nakagawa, N.; Kanda, E.; Nagasu, H.; Matsushita, K.; Nangaku, M.; Maruyama, S.; Wada, T.; Terada, Y.; Yamagata, K.; et al. Prevalences of hyperuricemia and electrolyte abnormalities in patients with chronic kidney disease in Japan: A nationwide, cross-sectional cohort study using data from the Japan Chronic Kidney Disease Database (J-CKD-DB). PLoS ONE 2020, 15, 0240402. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.P.; McLeod, M.L.; Robinson, R.R. An extrarenal mechanism for the maintenance of potassium balance in sever chronic renal failure. Trans. Assoc. Am. Phys. 1967, 80, 207–216. [Google Scholar]
- Sandle, G.I.; Gaiger, E.; Tapster, S.; Goodshep, T.H.J. Enhanced rectal potassium secretion in chronic renal insufficiency: Evidence for large intestinal potassium adaptation in man. Clin. Sci. 1986, 71, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Panese, S.; Mártin, R.S.; Virginillo, M.; Litardo, M.; Siga, E.; Arrizurieta, E.; Hayslett, J.P. Mechanism of enhanced transcellular potassium–secretion in man with chronic renal failure. Kidney Int. 1987, 31, 1377–1382. [Google Scholar] [CrossRef]
- Sandle, G.I.; Gaiger, E.; Tapster, S.; Goodship, T.H.J. Evidence for large intestinal control of potassium homoeostasis in uraemic patients undergoing long-term dialysis. Clin. Sci. 1987, 73, 247–252. [Google Scholar] [CrossRef]
- Sandle, G.I.; Hunter, M. Apical potassium (BK) channels and enhanced potassium secretion in human colon. Qjm: Int. J. Med. 2009, 103, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Unwin, R.J.; Luft, F.C.; Shirley, D.G. Pathophysiology and management of hypokalemia: A clinical perspective. Nat. Rev. Nephrol. 2011, 7, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.R.; Ing, T.S.; Metcalfe-Gibson, A.; Wrong, O.M. The chemical composition of faeces in uraemia, as revealed by in-vivo faecal dialysis. Clin. Sci. 1968, 35, 35. [Google Scholar]
- Yosipovitch, G.; Reis, J.; Tur, E.; Blau, H.; Harell, D.; Morduchowicz, G.; Boner, G. Sweat electrolytes in patients with advanced renal failure. J. Lab. Clin. Med. 1994, 124, 808–812. [Google Scholar]
- Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. The sixth report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch. Intern. Med. 1997, 157, 2413–2446. [Google Scholar]
- WHO. Guideline: Potassium Intake for Adults and Children; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Rabelink, T.; Koomans, H.A.; Hené, R.J.; Mees, E.J.D. Early and late adjustment to potassium loading in humans. Kidney Int. 1990, 38, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, H.N.; Swenson, K.; Settach, G. Cardiac arrest due to oral potassium administration. Am. J. Med. 1975, 58, 139–142. [Google Scholar] [CrossRef]
- Imai, E.; Horio, M.; Watanabe, T.; Iseki, K.; Yamagata, K.; Hara, S.; Ura, N.; Kiyohara, Y.; Moriyama, T.; Ando, Y.; et al. Prevalence of chronic kidney disease in the Japanese general population. Clin. Exp. Nephrol. 2009, 13, 621–630. [Google Scholar] [CrossRef]
- Takaichi, K.; Takemoto, F.; Ubara, Y.; Mori, Y. Analysis of Factors Causing Hyperkalemia. Intern. Med. 2007, 46, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.M.; Zhan, M.; Hsu, V.D.; Walker, L.D.; Moen, M.F.; Seliger, S.L.; Weir, M.R.; Fink, J.C. The Frequency of Hyperkalemia and Its Significance in Chronic Kidney Disease. Arch. Intern. Med. 2009, 169, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M.; Appel, L.J.; Bakris, G.L.; Gassman, J.J.; Greene, T.; Kendrick, C.A.; Wang, X.; Lash, J.P.; Lewis, J.A.; Pogue, V.; et al. Risk of Hyperkalemia in Nondiabetic Patients with Chronic Kidney Disease Receiving Antihypertensive TherapyHyperkalemia in CKD Adults Using Antihypertensives. Arch. Intern. Med. 2009, 169, 1587–1594. [Google Scholar] [CrossRef]
- Kim, H.W.; Park, J.T.; Yoo, T.H.; Lee, J.; Chung, W.; Lee, K.B.; Chae, D.W.; Ahn, C.; Kang, S.W.; Choi, K.H.; et al. Urinary potassium excretion and progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2019, 14, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Kolasa, K.M. Dietary approaches to stop hypertension (DASH) in clinical practice: A primary care experience. Clin. Cardiol. 1999, 22, 16–22. [Google Scholar] [CrossRef]
- Korgaonkar, S.; Tilea, A.; Gillespie, B.W.; Kiser, M.; Eisele, G.; Finkelstein, F.; Kotanko, P.; Pitt, B.; Saran, R. Serum potassium and outcomes in CKD: Insights from the RRI-CKD cohort study. Clin. J. Am. Soc. Nephrol. 2010, 5, 762–769. [Google Scholar] [CrossRef]
- Miao, Y.; Dobre, D.; Heerspink, H.J.L.; Brenner, B.M.; Cooper, M.E.; Parving, H.H.; Shahinfar, S.; Grobbee, D.; Zeeuw, D.E. Increased serum potassium affects renal outcomes: A post hoc analysis of the Reduction of Endpoints in NIDDM with the Angiotesin ll Antagonist Losartan (RENAAL) trial. Diabetologia 2011, 54, 44–50. [Google Scholar] [CrossRef]
- Smyth, A.; Dunkler, D.; Gao, P.; Teo, K.K.; Yusuf, S.; O’Donnell, M.J.; Mann, J.F.; Clase, C.M. The relationship between estimated sodium and potassium excretion and subsequent renal outcomes. Kidney Int. 2014, 86, 1205–1212. [Google Scholar] [CrossRef]
- He, J.; Mills, K.T.; Appel, L.J.; Yang, W.; Chen, J.; Lee, B.T.; Rosas, S.E.; Porter, A.; Makos, G.; Weir, M.R.; et al. Urinary Sodium and Potassium Excretion and CKD Progression. J. Am. Soc. Nephrol. 2015, 27, 1202–1212. [Google Scholar] [CrossRef]
- Kalantar, Z.K.; Tortorici, A.R.; Cen, J.L.T.; Kamgar, M.; Lau, W.L.; Moradi, H.; Rhee, C.M.; Streja, E.; Kovesdy, C.P. Dietary restrictions in dialysis patients: Is there anything left to eat? Semin. Dial. 2015, 28, 159–168. [Google Scholar] [CrossRef]
- Roberts, S.B.; Fuss, P.; Heyman, M.B.; Evans, W.J.; Tsay, R.; Rasmussen, H.; Fiatarone, M.; Cortiella, J.; Dallal, G.E.; Young, V.R. Control of food intake in older men. JAMA 1994, 272, 1601–1606. [Google Scholar] [CrossRef]
- Lynch, K.E.; Lynch, R.; Curhan, G.C.; Brunelli, S.M. Prescribed Dietary Phosphate Restriction and Survival among Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2010, 6, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Shinaberger, C.S.; Greenland, S.; Kopple, J.D.; Wyck, D.V.; Mehrotra, R.; Kovesdy, C.P.; Kalantar-Zadeh, K. Is controlling Phosphorus by decreasing dietary protein intake beneficial or harmful in persons with chronic kidney disease? Am. J. Clin. Nutr. 2008, 88, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Greene, J.H.; Wingard, R.L.; A Parker, R.; Hakim, R.M. Spontaneous dietary protein intake during progression of chronic renal failure. J. Am. Soc. Nephrol. 1995, 6, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; HeimbUrger, O.; Lindholm, B.; Kaysen, G.A.; Bergström, J. Are there two types of malnutrition in chronic renal failure? Evidence for relationships between malnutrition inflammation and atherosclerosis (MIA syndrome). Nephrol. Dial. Transpl. 2000, 15, 953–960. [Google Scholar] [CrossRef]
- Fouque, D.; Kalantar-Zadeh, K.; Kopple, J.; Cano, N.; Chauveau, P.; Cuppari, L.; Franch, H.; Guarnieri, G.; Ikizler, T.; Kaysen, G.; et al. A proposed nomenclature and diagnostic criteria for protein–energy wasting in acute and chronic kidney disease. Kidney Int. 2008, 73, 391–398. [Google Scholar] [CrossRef]
- Klahr, S.; Levey, A.S.; Beck, G.J.; Caggiula, A.W.; Hunsicker, L.; Kusek, J.W.; Striker, G. The effects of dietary protein restriction and blood pressure control on the progression of chronic renal disease. Modification of diet in renal disease study group. N. Engl. J. Med. 1994, 330, 877–884. [Google Scholar] [CrossRef]
- Foley, R.N.; Gilbertson, D.T.; Murray, T.; Collins, A.J. Long Interdialytic Interval and Mortality among Patients Receiving Hemodialysis. N. Engl. J. Med. 2011, 365, 1099–1107. [Google Scholar] [CrossRef]
- Zhang, H.; Schaubel, D.E.; Kalbfleisch, J.D.; Bragg-Gresham, J.L.; Robinson, B.M.; Pisoni, R.L.; Canaud, B.; Jadoul, M.; Akiba, T.; Saito, A.; et al. Dialysis outcomes and analysis of practice patterns suggests the dialysis schedule affects day-of-week mortality. Kidney Int. 2012, 81, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Yamaoka, Y.; Nagai, M.; Nakaya, T.; Hanba, Y.; Shigematsu, T. Analysis of factors affecting the prognosis of a hemodialysis patient-Focusing on albumin and parameters of nutrition. Nihon Toseki Igakkai Zasshi 2010, 43, 453–460. [Google Scholar] [CrossRef]
- Johansen, K.L.; Zhang, R.; Huang, Y.; Chen, S.C.; Blagg, C.R.; Goldfarb-Rumyantzev, A.S.; Hoy, C.D.; Lockridge, R.S., Jr.; Miller, B.W.; Eggers, P.W.; et al. Survival and hospitalization among patients using nocturnal and short daily compared to conventional hemodialysis: A USRDS study. Kindney Int. 2009, 76, 984–990. [Google Scholar] [CrossRef]
- Feriani, M.; Ronco, C.; Greca, G.L. Replacement of Renal Function by Dialysis, 5th ed.; Horl, W.H., Koch, K.M., Lindsay, R.M., Winchester, J.F., Eds.; Kluwer Academic Publishers: Lancaster, UK, 2004; pp. 505–537. [Google Scholar]
- Krishnamurthy, V.M.R.; Wei, G.; Baird, B.C.; Murtaugh, M.; Chonchol, M.B.; Raphael, K.L.; Greene, T.; Beddhu, S. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. 2012, 81, 300–306. [Google Scholar] [CrossRef]
- Tomemori, H.; Hamamura, K.; Tabane, K. Interactive Effects of Sodium and Potassium on the Growth and Photosynthesis of Spinach and Komatsuna. Plant Prod. Sci. 2002, 5, 281–285. [Google Scholar] [CrossRef]
- Ogawa, A.; Eguchi, T.; Toyofuku, K. Cultivation Methods for Leafy Vegetables and Tomatoes with Low Potassium Content for Dialysis Patients. Environ. Control. Biol. 2012, 50, 407–414. [Google Scholar] [CrossRef]
- Renna, M.; Castellino, M.; Leoni, B.; Paradiso, V.M.; Santamaria, P. Microgreens Production with Low Potassium Content for Patients with Impaired Kidney Function. Nutrients 2018, 10, 675. [Google Scholar] [CrossRef] [PubMed]
- Asao, T.; Asaduzzaman; Mondal, F.; Tokura, M.; Adachi, F.; Ueno, M.; Kawaguchi, M.; Yano, S.; Ban, T. Impact of reduced potassium nitrate concentrations in nutrient solution on the growth, yield and fruit quality of melon in hydroponics. Sci. Hortic. 2013, 164, 221–231. [Google Scholar] [CrossRef]
- Mondal, F.; Asaduzzaman; Ueno, M.; Kawaguchi, M.; Yano, S.; Ban, T.; Tanaka, H.; Asao, T. Reduction of Potassium (K) Content in Strawberry Fruits through KNO3 Management of Hydroponics. Hortic. J. 2017, 86, 26–36. [Google Scholar] [CrossRef]
- Talukder, R.; Asaduzzaman; Ueno, M.; Kawaguchi, M.; Yano, S.; Ban, T.; Tanaka, H.; Asao, T. Low Potassium Content Vegetables Research for Chronic Kidney Disease Patients in Japan. Nephrol. Open J. 2016, 2, 1–8. [Google Scholar] [CrossRef]
- Iwahori, T.; Ueshima, H.; Torii, S.; Saito, Y.; Kondo, K.; Tanaka-Mizuno, S.; Arima, H.; Miura, K. Diurnal variation of urinary sodium-to-potassium ratio in free-living Japanese individuals. Hypertens. Res. 2017, 40, 658–664. [Google Scholar] [CrossRef]
- Ethier, J.H.; Kamel, K.S.; Magner, P.O.; Lemann, J., Jr.; Halperin, M.L. The transtubular potassium concentration in patients with hypokale-mia and hyperkalemia. Am. J. Kidney Dis. 1990, 15, 309–315. [Google Scholar] [CrossRef]
- Choi, M.J.; Ziyadeh, F.N. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J. Am. Soc. Nephrol. 2008, l9, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Zettle, R.M.; West, M.L.; Josse, R.G.; Richardson, R.M.; Marsden, P.A.; Halperin, M.L. Renal potassium handling during states of low al-dosterone bioactivity: A method to differentiate renal and non-renal causes. Am. J. Nephrol. 1987, 7, 360–366. [Google Scholar] [CrossRef]
- Kamel, K.S.; Halperin, M.L. Intrarenal urea recycling leads to a higher rate of renal excretion of potassium: An hypothesis with clinical implications. Curr. Opin. Nephrol. Hypertens. 2011, 20, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Karet, F.E. Mechanisms in hyperkalelnic renal tubular acidosis. J. Am. Soc. Nephrol. 2009, 20, 251–254. [Google Scholar] [CrossRef]
- Watson, M.; Abbott, K.; Yuan, C.M. Damned If You Do, Damned If You Don’t: Potassium Binding Resins in Hyperkalemia. Clin. J. Am. Soc. Nephrol. 2010, 5, 1723–1726. [Google Scholar] [CrossRef]
- Gardiner, G.W. Kayexalate (sodium polystyrene sulphonate) in sorbitol associated with intestinal necrosis in uremic patients. Can. J. Gastroenterol. 1997, 11, 573–577. [Google Scholar] [CrossRef]
- Rashid, A.; Hamilton, S.R. Necrosis of the Gastrointestinal Tract in Uremic Patients as a Result of Sodium Polystyrene Sulfonate (Kayexalate) in Sorbitol. Am. J. Surg. Pathol. 1997, 21, 60–69. [Google Scholar] [CrossRef]
- Stavros, F.; Yang, A.; Leon, A.; Nuttall, M.; Rasmussen, H.S. Characterization of structure and function of ZS-9, a K+ selective ion trap. PLoS ONE 2014, 9, e114686. [Google Scholar] [CrossRef]
- Greenberg, A. Hyperkalemia: Treatment options. Semin. Nephrol. 1998, 18, 46–57. [Google Scholar] [PubMed]
- Allon, M.; Takeshian, A.; Shanklin, N. Effect of insulin plus glucose infusion with or without epirlephrine on fasting hyperkalemia. Kidney Int. 1993, 43, 212–217. [Google Scholar] [CrossRef][Green Version]
- Hsueh, W.A.; Carlson, E.J.; Luetscher, J.A.; Grislis, G. Activation and Characterization of Inactive Big Renin in Plasma of Patients with Diabetic Nephropathy and Unusual Active Renin*. J. Clin. Endocrinol. Metab. 1980, 51, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Mount, D.B. Disorders of potassium balance. In Brenner and Rector’s the Kidney, 10th ed.; Skorecki, K., Chertow, G.M., Marsden, P.A., Taal, M.W., Wasser, W.G., Eds.; Elsevier: Philadelphia, PA, USA, 2016; pp. 559–600. [Google Scholar]
- Palevsky, P.M.; Zhang, J.H.; O’Connor, T.Z.; Chertow, G.M.; Crowley, S.T.; Choudhury, D.; Finkel, K.W.; Kellum, J.A.; Paganini, E.P.; Schein, R.M.H.; et al. Intensity of Renal Support in Critically Ill Patients with Acute Kidney Injury. N. Engl. J. Med. 2008, 359, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Cass, A.; Norton, R.; Gallagher, M.; Su, S.; Cole, L.; Finfer, S.; McArthur, C.; McGuinness, S.; Myburgh, J.; et al. Intensity of Continuous Renal-Replacement Therapy in Critically Ill Patients. N. Engl. J. Med. 2009, 361, 1627–1638. [Google Scholar] [CrossRef]
- Luo, J.; Brunelli, S.M.; Jensen, D.E.; Yang, A. Association between Serum Potassium and Outcomes in Patients with Reduced Kidney Function. Clin. J. Am. Soc. Nephrol. 2015, 11, 90–100. [Google Scholar] [CrossRef]
- Pun, P.H.; Lehrich, R.W.; Honeycutt, E.F.; Herzog, C.A.; Middleton, J.P. Modifiable risk factors associated with sudden cardiac arrest within hemodialysis clinics. Kidney Int. 2011, 79, 218–227. [Google Scholar] [CrossRef]
- Diercks, D.B.; Shumaik, G.M.; A Harrigan, R.; Brady, W.J.; Chan, T.C. Electrocardiographic manifestations: Electrolyte abnormalities. J. Emerg. Med. 2004, 27, 153–160. [Google Scholar] [CrossRef]
- Mujais, S.K.; Katz, A.L. Potassium deficiency. In The Kidney: Physiology and Pathophysiology; Seldin, D.W., Giebisch, G., Eds.; Lippincott Wiliams & Wilkins: Philadelphia, PA, USA, 2000; p. 1615. [Google Scholar]
- Schwartz, W.B.; Relman, A.S. Effects of electrolyte disorders on renal structure and function. N. Engl. J. Med. 1967, 276, 383–389. [Google Scholar] [CrossRef]
- Cremer, W.; Bock, K.D. Symptoms and course of chronic hypokalemic nephropathy in man. Clin. Nephrol. 1977, 7, 112–119. [Google Scholar]
- Torres, V.E.; Young, W.F.; Offord, K.P.; Hattery, R.R. Association of Hypokalemia, Aldosteronism, and Renal Cysts. N. Engl. J. Med. 1990, 322, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, D.; Naruo, T.; Taguchi, S.; Umekita, Y.; Yoshida, H.; Nozoe, S. “End-stage kidney” in longstanding bulimia nervosa. Int. J. Eat. Disord. 2005, 38, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-L.; Kuo, E. Mechanism of Hypokalemia in Magnesium Deficiency. J. Am. Soc. Nephrol. 2007, 18, 2649–2652. [Google Scholar] [CrossRef] [PubMed]
- Ruml, L.A.; Pak, C.Y. Effect of potassium magnesium citrate on thiazide-induced hypokalemia and magnesium loss. Am. J. Kidney Dis. 1999, 34, 107–113. [Google Scholar] [CrossRef]
- Koeppen, B.M. The kidney and acid-base regulatio. Adv. Physiol. Educ. 2009, 33, 275–281. [Google Scholar] [CrossRef]
- Kraut, J.A.; Kurtz, I. Metabolic Acidosis of CKD: Diagnosis, Clinical Characteristics, and Treatment. Am. J. Kidney Dis. 2005, 45, 978–993. [Google Scholar] [CrossRef]
- Terker, A.S.; Zhang, C.; McCormick, J.A.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metab. 2015, 21, 39–50. [Google Scholar] [CrossRef]
- Xu, N.; Hirohama, D.; Ishizawa, K.; Chang, W.X.; Shimosawa, T.; Fujita, T.; Uchida, S.; Shibata, S. Hypokalemia and Pendrin Induction by Aldosterone. Hypertension 2017, 69, 855–862. [Google Scholar] [CrossRef]
- Palygin, O.; Levchenko, V.; Ilatovskaya, D.V.; Pavlov, T.S.; Pochynyuk, O.M.; Jacob, H.J.; Geurts, A.M.; Hodges, M.R.; Staruschenko, A. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight 2017, 2, e92331. [Google Scholar] [CrossRef]
- Kempner, W. Some effects of the rice diet treatment of kidney disease and hypertension. Bull. N. Y. Acad. Med. 1946, 22, 358–370. [Google Scholar]
- Kawano, Y.; Omae, T. Lifestyle modifications in the management of hypertension benefits and limitations. CVD Prev. 1998, 1, 336–346. [Google Scholar]
- Stamler, J.; Chan, Q.; Daviglus, M.L.; Dyer, A.R.; Horn, L.V.; Garside, D.B.; Miura, K.; Wu, Y.; Ueshima, H.; Zhao, L.; et al. Relation of dietary sodium (salt) to blood pressure and its possible modulation by other dietary factors: The INTERMAP study. Hypertension 2018, 71, 631–637. [Google Scholar] [CrossRef]
- Whelton, P.K.; He, J.; Cutler, J.A.; Brancati, F.L.; Appel, L.J.; Follmann, D.; Klag, M.J. Effects of oral potassium on blood pressure meta-analysis of randomized controlled clinical trials. JAMA 1997, 277, 1624–1632. [Google Scholar] [CrossRef]
- Akita, S.; Sacks, F.M.; Svetkey, L.P.; Conlin, P.R.; Kimura, G. DASH-sodium trial collaborative research group: Effects of the dietary approaches to stop hypertension (DASH) diet on the pressure-natriuresis relationship. Hypertension 2003, 42, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Colin, P.R.; Miller, E.R.; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Hedayati, S.S.; Minhajuddin, A.T.; Ijaz, A.; Moe, O.W.; Elsayed, E.F.; Reilly, R.F.; Huang, C.-L. Association of Urinary Sodium/Potassium Ratio with Blood Pressure: Sex and Racial Differences. Clin. J. Am. Soc. Nephrol. 2011, 7, 315–322. [Google Scholar] [CrossRef]
- Araki, S.-I.; Haneda, M.; Koya, D.; Kondo, K.; Tanaka, S.; Arima, H.; Kume, S.; Nakazawa, J.; Chin-Kanasaki, M.; Ugi, S.; et al. Urinary Potassium Excretion and Renal and Cardiovascular Complications in Patients with Type 2 Diabetes and Normal Renal Function. Clin. J. Am. Soc. Nephrol. 2015, 10, 2152–2158. [Google Scholar] [CrossRef]
- Macdonald, J.E.; Struthers, A.D. Optimal serum potassium level in cardiovascular patients? J. Am. Coll Cardiol. 2004, 43, 155–161. [Google Scholar] [CrossRef]
- Wang, H.-H.; Hung, C.-C.; Kuo, M.-C.; Chiu, Y.-W.; Chang, J.-M.; Tsai, J.-C.; Seifter, J.L.; Chen, H.-C.; Hwang, D.-Y.; Hwang, S.-J. Hypokalemia, Its Contributing Factors and Renal Outcomes in Patients with Chronic Kidney Disease. PLoS ONE 2013, 8, e67140. [Google Scholar] [CrossRef] [PubMed]
- Tyson, C.C.; Lin, P.H.; Corsino, L.; Batch, B.C.; Allen, J.; Sapp, S.; Barnhart, H.; Nwankwo, C.; Burroughs, J.; Svetkey, L.P. Short-term effects of the DASH diet in adults with moderate chronic kidney disease: A pilot feeding study. Clin. Kidney J. 2016, 9, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, T.; Miura, K.; Ueshima, H. Time to Consider Use of the Sodium-to-Potassium Ratio for Practical Sodium Reduction and Potassium Increase. Nutrients 2017, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, T.; Ueshima, H.; Miyagawa, N.; Ohgami, N.; Yamashita, H.; Ohkubo, T.; Murakami, Y.; Shiga, T.; Miura, K. Six random specimens of daytime casual urine on different days are sufficient to estimate daily sodium/potassium ratio in comparison to 7-day 24-h urine collections. Hypertens. Res. 2014, 37, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, T.; Ueshima, H.; Torii, S.; Saito, Y.; Fujiyoshi, A.; Ohkubo, T.; Miura, K. Four to seven random casual urine specimens are sufficient to estimate 24-h urinary sodium/potassium ratio in individuals with high blood pressure. J. Hum. Hypertens. 2016, 30, 328–334. [Google Scholar] [CrossRef]
- Okuyama, Y.; Uchida, H.A.; Iwahori, T.; Segawa, H.; Kato, A.; Takeuchi, H.; Kakio, Y.; Umebayashi, R.; Kitagawa, M.; Sugiyama, H.; et al. The relationship between repeated measurement of casual and 24-h urinary sodium-to-potassium ratio in patients with chronic kidney disease. J. Hum. Hypertens. 2018, 33, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F. Managing hyperkalemia caused by inhibitors of the renin angiotensin al dosterone system. N. Engl. J. Med. 2004, 351, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Chaker, B.S.; Atef, B.; Neila, F.; Raoudha, S.; Houssem, H. Drugoinduced hyperkalemia. Drug Saf. 2014, 37, 677–692. [Google Scholar]
- Veltri, K.T.; Mason, C. Medication-Induced Hypokalemia. Pharm. Ther. 2015, 40, 185–190. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Deficiency | Toxicity |
|---|---|
| Gastrointestinal symptoms | Neuromuscular symptoms |
| vomiting/anorexia | lip numbness |
| ileus | muscle weakness |
| Neuromuscular symptoms | dyspnea due to respiratory paralysis |
| tetraplegia | Arrhythmia |
| muscle weakness | bradycardia |
| dyspnea due to respiratory paralysis | ventricular fibrillation |
| Impaired insulin secretion | ventricular flutter |
| Kidney disorders | cardiac arrest |
| impaired urine concentration | Lassitude |
| tubulointerstitial changes | Loss of consciousness |
| Arrhythmia | |
| extrasystoles | |
| tachyarrhythmias | |
| atrioventricular block | |
| ventricular fibrillation | |
| Lassitude | |
| Loss of consciousness |
| All HD Patients (n = 42) | Non-DM Group (n = 20) | T2DM Group (n = 22) | p Value | |
|---|---|---|---|---|
| Age, years | 65.5 ± 11.1 | 66.1 ± 10.7 | 65.0 ± 11.7 | 0.6960 |
| Gender, male/female | 29/13 | 17/5 | 12/8 | 0.2322 |
| BMI, kg/m2 | 22.7 ± 4.5 | 21.3 ± 4.3 | 24.0 ± 4.3 | 0.0494 |
| HD duration, years | 4.6 (2.8–6.4) | 6.1 (3.4–8.0) | 4.2 (2.1–5.5) | 0.0365 |
| Interdialytic BW gain, % | 5.2 (4.3–5.8) | 5.3 (4.5–6.9) | 5.1 (4.3–5.5) | 0.2733 |
| Serum urea nitrogen, mg/dL | 61.3 ± 15.3 | 65.0 ± 17.3 | 58.0 ± 12.8 | 0.1511 |
| Cre, mg/dL | 10.0 ± 2.5 | 10.3 ± 2.7 | 10.5 ± 2.5 | 0.7818 |
| Alb, g/dL | 3.7 (3.4–3.8) | 3.5 (3.2–3.7) | 3.7 (3.5–4.0) | 0.0298 |
| Casual plasma glucose, mg/dL | 121.0 (101.0–149.0) | 107.0 (93.0–127.5) | 141.0 (117.0–162.0) | 0.0048 |
| Glycoalbumin, % | 16.6 ± 3.0 | 14.9 ± 2.1 | 18.1 ± 3.0 | 0.0004 |
| Na, mEq/L | 139.8 ± 3.4 | 139.3 ± 4.5 | 140.2 ± 2.2 | 0.6935 |
| K, mEq/L | 4.9 ± 0.7 | 5.1 ± 0.8 | 4.8 ± 0.7 | 0.2775 |
| Cl, mEq/L | 105.9 ± 3.3 | 105.6 ± 4.1 | 106.2 ± 2.4 | 0.7039 |
| PCR, g/day | 45.72 (42.09–53.29) | 45.65 (42.14–52.51) | 46.38 (40.88–53.29) | >0.9999 |
| n-PCR, g/kg/day | 0.834 ± 0.181 | 0.911 ± 0.185 | 0.765 ± 0.149 | 0.0135 |
| pH | 7.34 (7.33–7.36) | 7.34 (7.32–7.37) | 7.34 (7.33–7.36) | 0.5371 |
| HCO3, mEq/L | 19.8 ± 2.3 | 19.2 ± 2.7 | 20.4 ± 1.8 | 0.1658 |
| AcAc, µmol/L | 25.0 (21.0–44.0) | 24.5 (19.0–36.5) | 27.0 (22.0–53.0) | 0.2360 |
| β-HB, µmol/L | 20.5 (15.0–40.0) | 17.0 (12.5–33.0) | 30.5 (19.0–64.0) | 0.0070 |
| AcAc /β-HB ratio, µmol/µmol | 1.19 (0.75–1.47) | 1.35 (1.07–1.79) | 0.97 (0.69–1.24) | 0.0136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, S.; Inaba, M. Potassium Metabolism and Management in Patients with CKD. Nutrients 2021, 13, 1751. https://doi.org/10.3390/nu13061751
Yamada S, Inaba M. Potassium Metabolism and Management in Patients with CKD. Nutrients. 2021; 13(6):1751. https://doi.org/10.3390/nu13061751
Chicago/Turabian StyleYamada, Shinsuke, and Masaaki Inaba. 2021. "Potassium Metabolism and Management in Patients with CKD" Nutrients 13, no. 6: 1751. https://doi.org/10.3390/nu13061751
APA StyleYamada, S., & Inaba, M. (2021). Potassium Metabolism and Management in Patients with CKD. Nutrients, 13(6), 1751. https://doi.org/10.3390/nu13061751