
Nutrigenomics and Nutrigenetics in Metabolic- (Dysfunction) Associated Fatty Liver Disease: Novel Insights and Future Perspectives

 ,
,  ,
,  ,
,  ,
, 

Abstract

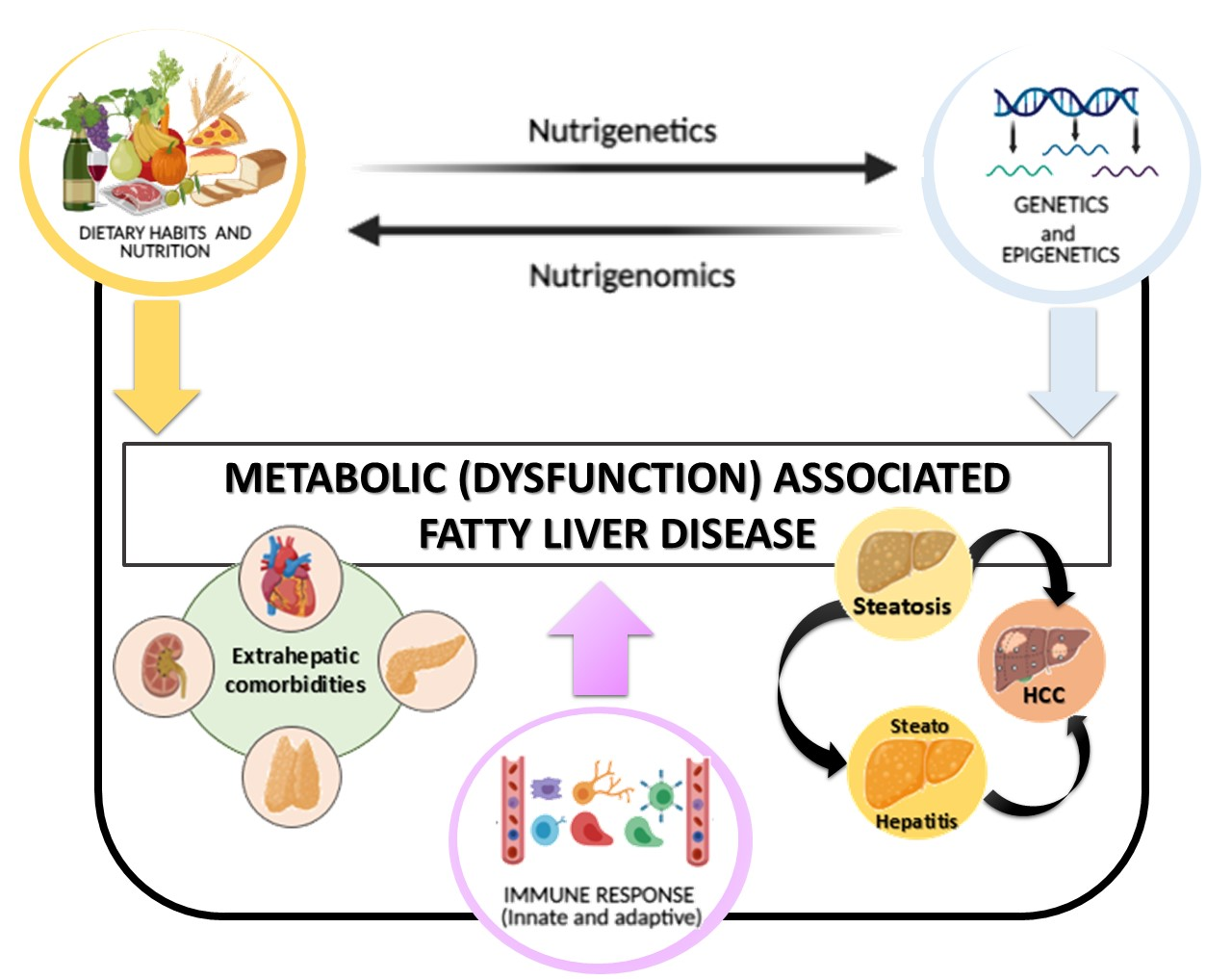
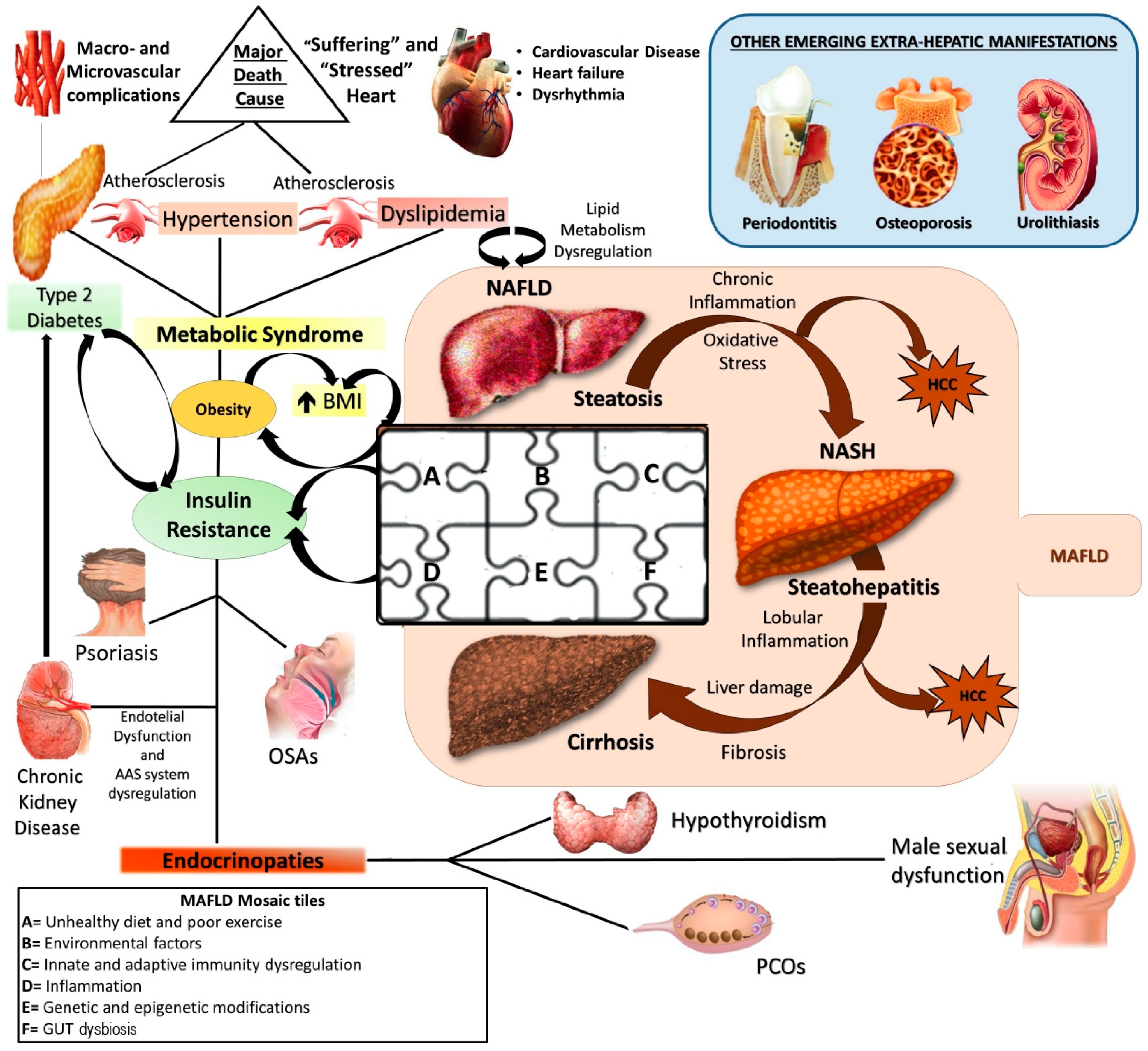
1. Introduction
2. Genetics and Epigenetics: An Overview on MAFLD Genetic Background
2.1. Most Common Genetic Determinants of MAFLD
2.2. Main Epigenetic Mechanisms of MAFLD
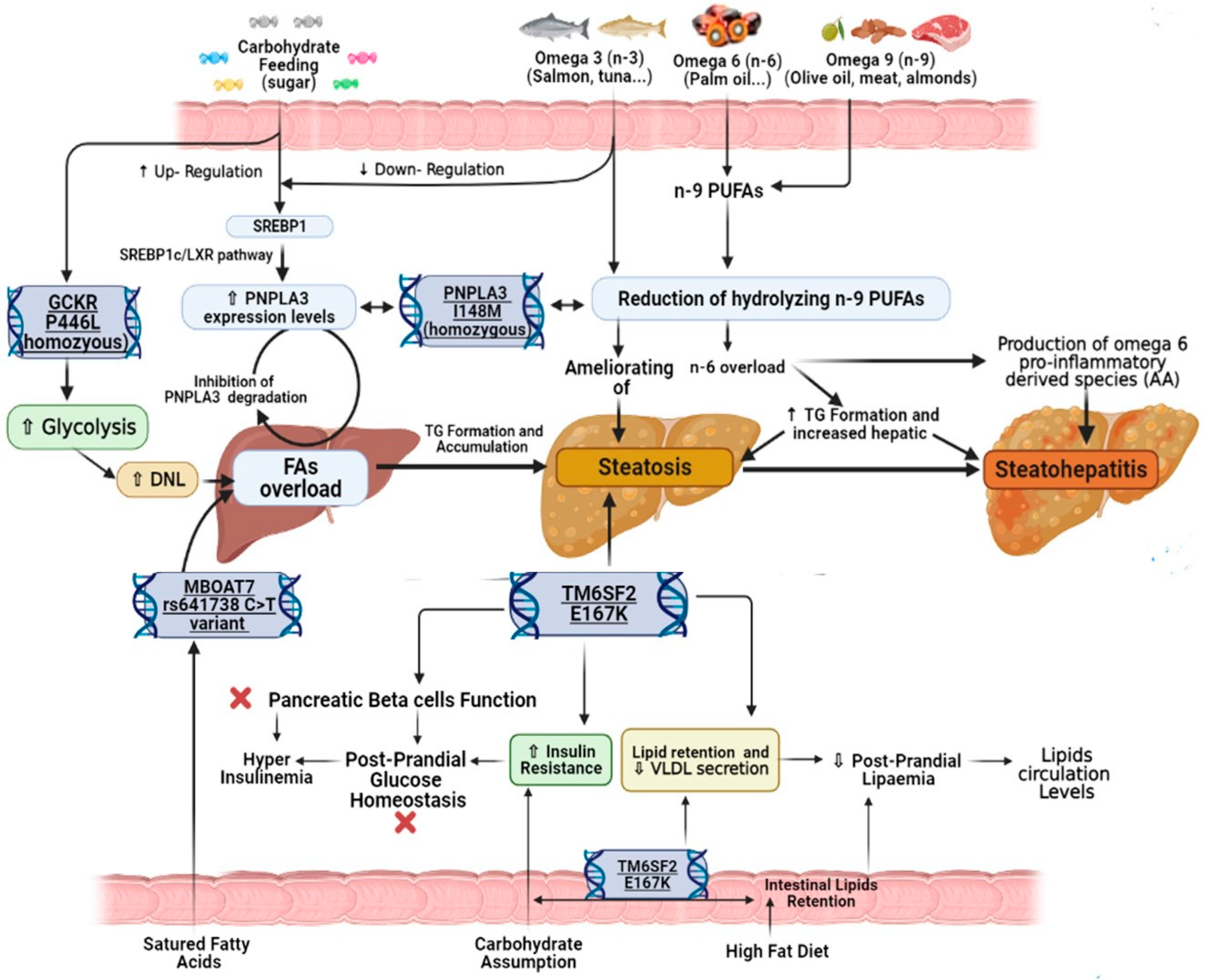
3. Nutrigenomics in MAFLD: The Impact of Nutrients on Epigenetic Regulation
3.1. Lipids
3.2. Carbohydrates
3.3. Proteins
3.4. Micronutrients
3.5. On the Way of a Genetic Based Dietary Approach
4. Nutrigenetics in MAFLD: Genetics Influence the Response to the Nutrients
4.1. PNPLA3 rs738409 Variant’s Influence
4.2. MBOAT7 rs641738 Variant’s Influence
4.3. TM6SF2 rs58542926 and GCKR rs1260326 Variants’ Influence
4.4. Personalized Nutritional Strategies Targeting Specific Genetic Backgrounds
5. Future Perspectives: Diet, Immunity, and Genes Interaction, a Very Complex Network That Needs to Be Explored
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ACC | Acetyl-CoA carboxylase |
| ACDRP | Amish Complex Disease Research Program |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ASO | antisense oligonucleotides |
| ATG7 | Autophagy related 7 |
| CHO | Carbohydrate enriched |
| CKD | Chronic kidney disease |
| DNL | De novo lipogenesis |
| FAs | Fatty acids |
| FASN | Fatty acid synthetase |
| FFAs | Free fatty acids |
| GCKR | Glucokinase regulator |
| GWAS | Genome-wide association studies |
| GPR | G protein-coupled receptor |
| HCC | Hepatocellular carcinoma |
| HDCA3 | Histone deacetylases |
| HFD | High-fat diet |
| HFE | Homeostatic iron regulator |
| HFF% | Hepatic fat fraction percentage |
| HFS | High-fat-sucrose |
| HSCs | Hepatic stellate cells |
| IL | Interleukin |
| IR | Insulin resistance |
| LA | Linoleic acid |
| LDL | Low-density lipoproteins |
| LDL-R | Low density lipoprotein receptor |
| LKB1 | Liver kinase B1 (LKB1) |
| LOX-1 | Lectin-like oxidized LDL receptor-1 |
| LXR | Liver X receptor |
| MAFLD | Metabolic (dysfunction) associated fatty liver disease |
| MARC1 | Mitochondrial amidoxime reducing component 1 |
| MBOAT7 | Membrane-bound O-acyltransferase domain containing 7 |
| MD | Mediterranean diet |
| MiRNAs | MicroRNAs |
| MS | Metabolic syndrome |
| Mt-DNA | Mitochondrial DNA |
| MT-ND6 | Mitochondrially encoded NADH ubiquinone oxidoreductase core subunit 6 |
| mTOR | mammalian target of rapamycin |
| MTTP | Microsomal triglyceride transfer protein |
| n−3 | Omega 3 |
| n−6 | Omega 6 |
| n−9 | Omega 9 |
| NAFLD | Non-alcoholic fatty liver disease |
| NAS | NAFLD activity score |
| NASH | Non-alcoholic steatohepatitis |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR family pyrin domain containing 3 |
| OxLDL | oxidized low-density lipoproteins |
| PARVB variant 1 | Homo sapiens parvin beta transcript variant 1 |
| PCSK7 | Proprotein convertase subtilisin/Kexin type 7 |
| PDGF-α | Platelet derived growth factor-alpha |
| PGC1 | peroxisome proliferator-activated receptor -coactivator-1beta |
| PI | Phosphatidylinositol |
| PNPLA3 | Phospholipase domain-containing protein-3 |
| PPAR | Peroxisome proliferator-activated receptor |
| PPP1R3B | Protein phosphatase 1 regulatory subunit 3B |
| PUFAs | Polyunsaturated fatty acids |
| ROS | Reactive oxygen species |
| SCFAs | short chain fatty acids |
| SIRT | Sirtuin |
| Sirtuins | Silent information regulator 2 proteins |
| SNP | Single nucleotide polymorphism |
| SOD 2 | Superoxide dismutase 2 |
| SR-A | Scavenger receptor A |
| SREBP1 | Sterol regulatory element-binding proteins |
| SsRNAs | Small non-coding single strand RNAs |
| TG | Triglycerides |
| TGF-β | Transforming growth factor-beta |
| TI | Trained immunity |
| TLRs | Toll like receptors |
| TM6SF2 | Transmembrane 6 superfamily member 2 protein |
| TNF-α | Tumour necrosis factor-α |
| UCP2 | Uncoupling protein 2 |
| UCP3 | Uncoupling Protein 3 |
| UFAs | Unsaturated fatty acids |
| UTR | Untranslated region |
| VLDL | Very-low-density lipoproteins |
References
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pr. 2020, 35, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-L.; Chen, H.; Wang, C.-L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model. ” World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755. [Google Scholar] [CrossRef]
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef]
- Dallio, M.; Sangineto, M.; Romeo, M.; Villani, R.; Romano, A.D.; Loguercio, C.; Serviddio, G.; Federico, A. Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression. Int. J. Mol. Sci. 2021, 22, 436. [Google Scholar] [CrossRef]
- Bibbò, S.; Ianiro, G.; Dore, M.P.; Simonelli, C.; Newton, E.E.; Cammarota, G. Gut Microbiota as a Driver of Inflammation in Nonalcoholic Fatty Liver Disease. Mediat. Inflamm. 2018, 2018, 1–7. [Google Scholar] [CrossRef]
- Nicolucci, C.; Errico, S.; Federico, A.; Dallio, M.; Loguercio, C.; Diano, N. Human exposure to Bisphenol A and liver health status: Quantification of urinary and circulating levels by LC–MS/MS. J. Pharm. Biomed. Anal. 2017, 140, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Lama, S.; Vanacore, D.; Diano, N.; Nicolucci, C.; Errico, S.; Dallio, M.; Federico, A.; Loguercio, C.; Stiuso, P. Ameliorative effect of Silybin on bisphenol A induced oxidative stress, cell proliferation and steroid hormones oxidation in HepG2 cell cultures. Sci. Rep. 2019, 9, 3228. [Google Scholar] [CrossRef]
- Eslam, M.; Sarin, S.K.; Wong, V.W.-S.; Fan, J.-G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.-H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 889–919. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L. A Nutrigenomic Approach to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2017, 18, 1534. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Noce, A.; Bigioni, M.; Calabrese, V.; Della Rocca, D.; Daniele, N.; Tozzo, C.; Renzo, L. The Effects of Italian Mediterranean Organic Diet (IMOD) on Health Status. Curr. Pharm. Des. 2010, 16, 814–824. [Google Scholar] [CrossRef]
- Davis, C.R.; Bryan, J.; Hodgson, J.M.; Murphy, K.J. Definition of the Mediterranean Diet: A Literature Review. Nutrients 2015, 7, 9139–9153. [Google Scholar] [CrossRef]
- Kim, K.; Hyeon, J.; Lee, S.A.; Kwon, S.O.; Lee, H.; Keum, N.; Lee, J.; Park, S.M. Role of Total, Red, Processed, and White Meat Consumption in Stroke Incidence and Mortality: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. J. Am. Hear. Assoc. 2017, 6, e005983. [Google Scholar] [CrossRef]
- Abenavoli, L.; Boccuto, L.; Federico, A.; Dallio, M.; Loguercio, C.; Di Renzo, L.; De Lorenzo, A. Diet and Non-Alcoholic Fatty Liver Disease: The Mediterranean Way. Int. J. Environ. Res. Public Health 2019, 16, 3011. [Google Scholar] [CrossRef]
- Dayeh, T.A.; Olsson, A.H.; Volkov, P.; Almgren, P.; Rönn, T.; Ling, C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia 2013, 56, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and Genetics in NAFLD: The Perfect Binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Ferguson, L.R.; De Caterina, R.; Görman, U.; Allayee, H.; Kohlmeier, M.; Prasad, C.; Choi, M.S.; Curi, R.; De Luis, D.A.; Gil, Á.; et al. Guide and Position of the International Society of Nutrigenetics/Nutrigenomics on Personalised Nutrition: Part 1—Fields of Precision Nutrition. Lifestyle Genom. 2016, 9, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Long, M.T.; Gurary, E.B.; Massaro, J.M.; Ma, J.; Hoffmann, U.; Chung, R.T.; Benjamin, E.J.; Loomba, R. Parental non-alcoholic fatty liver disease increases risk of non-alcoholic fatty liver disease in offspring. Liver Int. 2019, 39, 740–747. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Sánchez-Torrijos, Y.; Herrojo, J.A.; Palacios, D.P.; Gallego-Durán, R.; Romero-Gómez, M.; Ampuero, J. Analysis of the burden and variability in the management of NAFLD patients in the clinical practice: Unifying the required criteria. Rev. Española Enferm. Dig. 2019, 111, 270–274. [Google Scholar] [CrossRef]
- Rinella, M.E.; Tacke, F.; Sanyal, A.J.; Anstee, Q.M.; the Participants of the AASLD/EASL Workshop. Report on the AASLD/EASL Joint Workshop on Clinical Trial Endpoints in NAFLD. Hepatology 2019, 70, 1424–1436. [Google Scholar] [CrossRef]
- Clarke, J.D.; Cherrington, N.J. Nonalcoholic steatohepatitis in precision medicine: Unraveling the factors that contribute to individual variability. Pharmacol. Ther. 2015, 151, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Duseja, A. Genetic and epigenetic disease modifiers: Non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y. Genetic and epigenetic variants influencing the development of nonalcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 6546–6551. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; A Pennacchio, L.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 574–580. [Google Scholar] [CrossRef]
- Rae-Whitcombe, S.M.; Kennedy, D.; Voyles, M.; Thompson, M.P. Regulation of the promoter region of the human adiponutrin/PNPLA3 gene by glucose and insulin. Biochem. Biophys. Res. Commun. 2010, 402, 767–772. [Google Scholar] [CrossRef]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef] [PubMed]
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef]
- Haaker, M.W.; Vaandrager, A.B.; Helms, J.B. Retinoids in health and disease: A role for hepatic stellate cells in affecting retinoid levels. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158674. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Gambino, R. PNPLA3 rs738409 and TM6SF2 rs58542926 gene variants affect renal disease and function in nonalcoholic fatty liver disease. Hepatology 2015, 62, 658–659. [Google Scholar] [CrossRef]
- Murphy, R.C.; Folco, G. Lysophospholipid acyltransferases and leukotriene biosynthesis: Intersection of the Lands cycle and the arachidonate PI cycle. J. Lipid Res. 2019, 60, 219–226. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265. [Google Scholar] [CrossRef]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; Hooft, F.V. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.G.; McPherson, S.W.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Haridas, P.N.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Jackson, A.U.; Li, Y.K.; Stringham, H.M.; Kuusisto, J.; Kangas, A.J.; Soininen, P.; Ala-Korpela, M.; Burant, C.F.; Salomaa, V.; et al. Novel association of TM6SF2 rs58542926 genotype with increased serum tyrosine levels and decreased apoB-100 particles in Finns. J. Lipid Res. 2017, 58, 1471–1481. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Männistö, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef]
- Namikawa, C.; Shu-Ping, Z.; Vyselaar, J.R.; Nozaki, Y.; Nemoto, Y.; Ono, M.; Akisawa, N.; Saibara, T.; Hiroi, M.; Enzan, H.; et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 2004, 40, 781–786. [Google Scholar] [CrossRef]
- Macaluso, F.S.; Maida, M.; Petta, S. Genetic background in nonalcoholic fatty liver disease: A comprehensive review. World J. Gastroenterol. 2015, 21, 11088–11111. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE Genotype, Parenchymal Iron Accumulation, and Liver Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; A De Luis, D.; Izaola, O.; Sagrado, M.G.; Conde, R.; Alvarez, T.; Pacheco, D.; Velasco, M.C. Role of -55CT polymorphism of UCP3 gene on non alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572–576. [Google Scholar]
- Fares, R.; Petta, S.; Lombardi, R.; Grimaudo, S.; Dongiovanni, P.; Pipitone, R.; Rametta, R.; Fracanzani, A.L.; Mozzi, E.; Craxì, A.; et al. The UCP2 -866 G>A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. 2015, 35, 1574–1580. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Baselli, G.; Mancina, R.M.; Ruscica, M.; Longo, M.; Rametta, R.; Cespiati, A.; Pelusi, S.; Ferri, N.; et al. PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. J. Lipid Res. 2019, 60, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-TruncatingHSD17B13Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Mao, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Gustafsson, J.-A.; Guan, Y. Role of HSD17B13 in the liver physiology and pathophysiology. Mol. Cell. Endocrinol. 2019, 489, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of The Liver; European Association for the Study of Diabetes. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Msc, A.P.C.; Carver, T.; Hartley, S.; De Villiers, C.B.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef]
- Pelusi, S.; Baselli, G.; Pietrelli, A.; Dongiovanni, P.; Donati, B.; McCain, M.V.; Meroni, M.; Fracanzani, A.L.; Romagnoli, R.; Petta, S.; et al. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef]
- Guzman, C.B.; Duvvuru, S.; Akkari, A.; Bhatnagar, P.; Battioui, C.; Foster, W.; Zhang, X.M.; Shankar, S.S.; Deeg, M.A.; Chalasani, N.; et al. Coding variants in PNPLA3 and TM6SF2 are risk factors for hepatic steatosis and elevated serum alanine aminotransferases caused by a glucagon receptor antagonist. Hepatol. Commun. 2018, 2, 561–570. [Google Scholar] [CrossRef]
- Scorletti, E.; Bhatia, L.; McCormick, K.; Clough, G.; Nash, K.; Calder, P.; Byrne, C.D. Design and rationale of the WELCOME trial: A randomised, placebo controlled study to test the efficacy of purified long chain omega-3 fatty treatment in non-alcoholic fatty liver disease. Contemp. Clin. Trials 2014, 37, 301–311. [Google Scholar] [CrossRef]
- Sun, C.; Fan, J.-G.; Qiao, L. Potential Epigenetic Mechanism in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2015, 16, 5161–5179. [Google Scholar] [CrossRef]
- Hotta, K.; Kitamoto, A.; Kitamoto, T.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of differentially methylated region (DMR) networks associated with progression of nonalcoholic fatty liver disease. Sci. Rep. 2018, 8, 13567. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Imajo, K.; Saito, S.; Yoneda, M.; Nakamura, T.; Nakajima, A.; Hotta, K. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley–Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between Methylome and Transcriptome in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 2015, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Tang, D.; Du, Y.L.; Cao, C.Y.; Nie, Y.Q.; Cao, J.; Zhou, Y.J. Fatty liver mediated by peroxisome proliferator-activated receptor-α DNA methylation can be reversed by a methylation inhibitor and curcumin. J. Dig. Dis. 2018, 19, 421–430. [Google Scholar] [CrossRef]
- Chen, H.-C.; Chen, Y.-Z.; Wang, C.-H.; Lin, F.-J. The nonalcoholic fatty liver disease-like phenotype and lowered serum VLDL are associated with decreased expression and DNA hypermethylation of hepatic ApoB in male offspring of ApoE deficient mothers fed a with Western diet. J. Nutr. Biochem. 2020, 77, 108319. [Google Scholar] [CrossRef]
- Tyagi, S.; Sharma, S.; Gupta, P.; Saini, A.S.; Kaushal, C. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- De Mello, V.D.; Matte, A.; Perfilyev, A.; Männistö, V.; Rönn, T.; Nilsson, E.; Käkelä, P.; Ling, C.; Pihlajamäki, J. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017, 12, 287–295. [Google Scholar] [CrossRef]
- de Mello, V.D.; Sehgal, R.; Männistö, V.; Klåvus, A.; Nilsson, E.; Perfilyev, A.; Kaminska, D.; Miao, Z.; Pajukanta, P.; Ling, C.; et al. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. 2021, 41, 754–763. [Google Scholar] [CrossRef]
- Saran, A.R.; Dave, S.; Zarrinpar, A. Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease. Gastroenterology 2020, 158, 1948–1966.e1. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Vinciguerra, M.; Oben, J.; Tarquini, R.; De Cosmo, S. Non-alcoholic fatty liver disease: The role of nuclear receptors and circadian rhythmicity. Liver Int. 2014, 34, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; A Ibdah, J. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; O Castaño, G.; Sookoian, S. Liver mitochondrial DNA damage and genetic variability of Cytochrome b—A key component of the respirasome—Drive the severity of fatty liver disease. J. Intern. Med. 2021, 289, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef]
- Yoshino, J.; Imai, S.-I. Mitochondrial SIRT3: A New Potential Therapeutic Target for Metabolic Syndrome. Mol. Cell 2011, 44, 170–171. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef]
- Finch, M.L.; Marquardt, J.U.; Yeoh, G.C.; Callus, B.A. Regulation of microRNAs and their role in liver development, regeneration and disease. Int. J. Biochem. Cell Biol. 2014, 54, 288–303. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Long, J.-K.; Dai, W.; Zheng, Y.-W.; Zhao, S.-P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Salvoza, N.C.; Klinzing, D.C.; Gopez-Cervantes, J.; Baclig, M.O. Association of Circulating Serum miR-34a and miR-122 with Dyslipidemia among Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0153497. [Google Scholar] [CrossRef]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Salinas, D.C.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed]
- Erhartova, D.; Cahova, M.; Dankova, H.; Heczkova, M.; Mikova, I.; Sticova, E.; Spicak, J.; Seda, O.; Trunecka, P. Serum miR-33a is associated with steatosis and inflammation in patients with non-alcoholic fatty liver disease after liver transplantation. PLoS ONE 2019, 14, e0224820. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; De Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.S.; Lakhani, H.V.; Zehra, M.; Wang, J.; Dilip, A.; Puri, N.; O’Hanlon, K.; Sodhi, K. Predicting Nonalcoholic Fatty Liver Disease through a Panel of Plasma Biomarkers and MicroRNAs in Female West Virginia Population. Int. J. Mol. Sci. 2020, 21, 6698. [Google Scholar] [CrossRef]
- Hur, W.; Lee, J.H.; Kim, S.W.; Kim, J.-H.; Bae, S.H.; Kim, M.; Hwang, D.; Kim, Y.S.; Park, T.; Um, S.-J.; et al. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/AKT pathway. Int. J. Biochem. Cell Biol. 2015, 64, 265–276. [Google Scholar] [CrossRef]
- Liu, X.-L.; Pan, Q.; Zhang, R.-N.; Shen, F.; Yan, S.-Y.; Sun, C.; Xu, Z.-J.; Chen, Y.-W.; Fan, J.-G. Disease-specific miR-34a as diagnostic marker of non-alcoholic steatohepatitis in a Chinese population. World J. Gastroenterol. 2016, 22, 9844–9852. [Google Scholar] [CrossRef]
- Perakakis, N.; Stefanakis, K.; Mantzoros, C.S. The role of omics in the pathophysiology, diagnosis and treatment of non-alcoholic fatty liver disease. Metabolism 2020, 111, 154320. [Google Scholar] [CrossRef]
- Mullins, V.A.; Bresette, W.; Johnstone, L.; Hallmark, B.; Chilton, F.H. Genomics in Personalized Nutrition: Can You “Eat for Your Genes”? Nutrients 2020, 12, 3118. [Google Scholar] [CrossRef] [PubMed]
- Blasbalg, T.L.; Hibbeln, J.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef]
- Lottenberg, A.M.; Afonso, M.D.S.; Lavrador, M.S.F.; Machado, R.M.; Nakandakare, E.R. The role of dietary fatty acids in the pathology of metabolic syndrome. J. Nutr. Biochem. 2012, 23, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Lytle, K.A.; Depner, C.M.; Tripathy, S. Omega-3 polyunsaturated fatty acids as a treatment strategy for nonalcoholic fatty liver disease. Pharmacol. Ther. 2018, 181, 108–125. [Google Scholar] [CrossRef]
- Levy, J.R.; Clore, J.N.; Stevens, W. Dietary n-3 polyunsaturated fatty acids decrease hepatic triglycerides in Fischer 344 rats. Hepatology 2004, 39, 608–616. [Google Scholar] [CrossRef]
- Campos, H.; Baylin, A.; Willett, W.C. α-Linolenic Acid and Risk of Nonfatal Acute Myocardial Infarction. Circulation 2008, 118, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.C.; Ryan, A.S.; Hafer-Macko, C.E.; Yepes, M.; Nahab, F.B.; Ziegler, T.R. Dietary and Serum Omega-6/Omega-3 Fatty Acids Are Associated with Physical and Metabolic Function in Stroke Survivors. Nutrients 2020, 12, 701. [Google Scholar] [CrossRef]
- Lankinen, M.A.; Fauland, A.; Shimizu, B.-I.; Ågren, J.; E Wheelock, C.; Laakso, M.; Schwab, U.; Pihlajamäki, J. Inflammatory response to dietary linoleic acid depends on FADS1 genotype. Am. J. Clin. Nutr. 2019, 109, 165–175. [Google Scholar] [CrossRef]
- Lankinen, M.A.; de Mello, V.D.; Meuronen, T.; Sallinen, T.; Ågren, J.; Virtanen, K.A.; Laakso, M.; Pihlajamäki, J.; Schwab, U. The FADS1 Genotype Modifies Metabolic Responses to the Linoleic Acid and Alpha-linolenic Acid Containing Plant Oils–Genotype Based Randomized Trial FADSDIET2. Mol. Nutr. Food Res. 2021, 65, e2001004. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, K.; Yu, W.; Wang, H.; Liu, L.; Wu, Q.; Li, S.; Guo, J. MiR-181b regulates steatosis in nonalcoholic fatty liver disease via targeting SIRT1. Biochem. Biophys. Res. Commun. 2017, 493, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Crudele, A.; Panera, N.; Romito, I.; Meroni, M.; De Stefanis, C.; Palma, A.; Comparcola, D.; Fracanzani, A.L.; Miele, L.; et al. β-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 2020, 72, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.-M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol Activation of Protein Kinase Cε and Hepatic Insulin Resistance. Cell Metab. 2012, 15, 574–584. [Google Scholar] [CrossRef]
- Santos-Marcos, J.A.; Perez-Jimenez, F.; Camargo, A. The role of diet and intestinal microbiota in the development of metabolic syndrome. J. Nutr. Biochem. 2019, 70, 1–27. [Google Scholar] [CrossRef]
- Ma, J.; Fox, C.S.; Jacques, P.F.; Speliotes, E.K.; Hoffmann, U.; Smith, C.E.; Saltzman, E.; McKeown, N.M. Sugar-sweetened beverage, diet soda, and fatty liver disease in the Framingham Heart Study cohorts. J. Hepatol. 2015, 63, 462–469. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.D.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; for the Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kast, P.; Böhnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mähler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Machann, J.; Küper, M.A.; Maier, I.B.; Spruss, A.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. A moderate weight reduction through dietary intervention decreases hepatic fat content in patients with non-alcoholic fatty liver disease (NAFLD): A pilot study. Eur. J. Nutr. 2013, 52, 527–535. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Jung, K.S.; Kim, S.U.; Yoon, H.-J.; Yun, Y.J.; Lee, B.-W.; Kang, E.S.; Han, K.-H.; Lee, H.C.; Cha, B.-S. Sarcopaenia is associated with NAFLD independently of obesity and insulin resistance: Nationwide surveys (KNHANES 2008–2011). J. Hepatol. 2015, 63, 486–493. [Google Scholar] [CrossRef]
- Wu, H.; Jang, J.; Dridi, S.; Ferrando, A.A.; Wolfe, R.R.; Kim, I.; Baum, J.I. Net protein balance correlates with expression of autophagy, mitochondrial biogenesis, and fat metabolism-related genes in skeletal muscle from older adults. Physiol. Rep. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Autophagy in skeletal muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 Ubiquitin Ligases and Mitophagy-Related Genes in Skeletal Muscle of Physically Inactive, Frail Older Women: A Cross-Sectional Comparison. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- Cordero, P.; Milagro, F.I.; Campion, J.; Martinez, J.A. Maternal methyl donors supplementation during lactation prevents the hyperhomocysteinemia induced by a high-fat-sucrose intake by dams. Int. J. Mol. Sci. 2013, 14, 24422–24437. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Zhang, H.-W.; Zhou, J.-Y.; Liu, Y.; Yang, Y.; Chen, X.-L.; Zhu, C.-H.; Zheng, R.-D.; Ling, W.-H.; Zhu, H.-L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD + and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Wang, C.; Liu, W.; Yao, L.; Zhang, X.; Zhang, X.; Ye, C.; Jiang, H.; He, J.; Zhu, Y.; Ai, D. Hydroxyeicosapentaenoic acids and epoxyeicosatetraenoic acids attenuate early occurrence of nonalcoholic fatty liver disease. Br. J. Pharmacol. 2017, 174, 2358–2372. [Google Scholar] [CrossRef]
- Shen, J.; Wong, G.L.-H.; Chan, H.L.-Y.; Chan, R.S.-M.; Chan, H.-Y.; Chu, W.C.-W.; Cheung, B.H.-K.; Yeung, D.K.-W.; Li, L.S.; Sea, M.M.-M.; et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2015, 30, 139–146. [Google Scholar] [CrossRef]
- Caulfield, T.; Ries, N.; Ray, P.; Shuman, C.; Wilson, B.; Ries, N. Direct-to-consumer genetic testing: Good, bad or benign? Clin. Genet. 2010, 77, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Heianza, Y.; Qi, L. Gene-Diet Interaction and Precision Nutrition in Obesity. Int. J. Mol. Sci. 2017, 18, 787. [Google Scholar] [CrossRef] [PubMed]
- Stover, P.J.; Caudill, M.A. Genetic and Epigenetic Contributions to Human Nutrition and Health: Managing Genome–Diet Interactions. J. Am. Diet. Assoc. 2008, 108, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Nick, A.; Hölttä-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Brück, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.; Lallukka-Brück, S.; Luukkonen, P.K.; Zhou, Y.; Gastaldelli, A.; Orho-Melander, M.; Sammalkorpi, H.; Juuti, A.; Penttilä, A.K.; Perttilä, J.; et al. The PNPLA3-I148M variant increases polyunsaturated triglycerides in human adipose tissue. Liver Int. 2020, 40, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef]
- Santoro, N.; Savoye, M.; Kim, G.; Marotto, K.; Shaw, M.M.; Pierpont, B.; Caprio, S. Hepatic Fat Accumulation Is Modulated by the Interaction between the rs738409 Variant in the PNPLA3 Gene and the Dietary Omega6/Omega3 PUFA Intake. PLoS ONE 2012, 7, e37827. [Google Scholar] [CrossRef]
- Innes, J.K.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48. [Google Scholar] [CrossRef]
- Bjermo, H.; Iggman, D.; Kullberg, J.; Dahlman, I.; Johansson, L.; Persson, L.; Berglund, J.; Pulkki, K.; Basu, S.; Uusitupa, M.; et al. Effects of n-6 PUFAs compared with SFAs on liver fat, lipoproteins, and inflammation in abdominal obesity: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 95, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Ingle, K.A.; Kachman, M.; Baum, H.; Shanmugam, G.; Rajasekaran, N.S.; Young, M.E.; Halade, G.V. Excess ω-6 fatty acids influx in aging drives metabolic dysregulation, electrocardiographic alterations, and low-grade chronic inflammation. Am. J. Physiol. Circ. Physiol. 2018, 314, H160–H169. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Van Name, M.A.; Savoye, M.; Chick, J.M.; Galuppo, B.T.; Feldstein, A.E.; Pierpont, B.; Johnson, C.; Shabanova, V.; Ekong, U.; Valentino, P.L.; et al. A Low ω-6 to ω-3 PUFA Ratio (n–6:n–3 PUFA) Diet to Treat Fatty Liver Disease in Obese Youth. J. Nutr. 2020, 150, 2314–2321. [Google Scholar] [CrossRef]
- Davis, J.N.; Lê, K.-A.; Walker, R.W.; Vikman, S.; Spruijt-Metz, N.; Weigensberg, M.J.; Allayee, H.; Goran, M.I. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am. J. Clin. Nutr. 2010, 92, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Liccardo, D.; Bedogni, G.; Salvatori, G.; Gnani, D.; Bersani, I.; Alisi, A.; Valenti, L.; Raponi, M. Influence of dietary pattern, physical activity, and I148M PNPLA3 on steatosis severity in at-risk adolescents. Genes Nutr. 2014, 9, 392. [Google Scholar] [CrossRef]
- Mater, M.K.; Thelen, A.P.; Pan, D.A.; Jump, D.B. Sterol Response Element-binding Protein 1c (SREBP1c) Is Involved in the Polyunsaturated Fatty Acid Suppression of Hepatic S14 Gene Transcription. J. Biol. Chem. 1999, 274, 32725–32732. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef]
- Ma, D.; Wang, Z.; Merrikh, C.N.; Lang, K.S.; Lu, P.; Li, X.; Merrikh, H.; Rao, Z.; Xu, W. Crystal structure of a membrane-bound O-acyltransferase. Nat. Cell Biol. 2018, 562, 286–290. [Google Scholar] [CrossRef]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef]
- O’Hare, E.A.; Yang, R.; Yerges-Armstrong, L.M.; Sreenivasan, U.; McFarland, R.; Leitch, C.C.; Wilson, M.H.; Narina, S.; Gorden, A.; Ryan, K.A.; et al. TM6SF2 rs58542926 impacts lipid processing in liver and small intestine. Hepatology 2017, 65, 1526–1542. [Google Scholar] [CrossRef]
- Musso, G.; Cipolla, U.; Cassader, M.; Pinach, S.; Saba, F.; De Michieli, F.; Paschetta, E.; Bongiovanni, D.; Framarin, L.; Leone, N.; et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J. Lipid Res. 2017, 58, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Franzago, M.; Fraticelli, F.; Nicolucci, A.; Celentano, C.; Liberati, M.; Stuppia, L.; Vitacolonna, E. Molecular Analysis of a Genetic Variants Panel Related to Nutrients and Metabolism: Association with Susceptibility to Gestational Diabetes and Cardiometabolic Risk in Affected Women. J. Diabetes Res. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; McKeown, N.M.; Kanoni, S.; Lemaitre, R.N.; Hivert, M.-F.; Ngwa, J.; Van Rooij, F.J.A.; Sonestedt, E.; Wojczynski, M.K.; Ye, Z.; et al. Interactions of Dietary Whole-Grain Intake With Fasting Glucose- and Insulin-Related Genetic Loci in Individuals of European Descent: A meta-analysis of 14 cohort studies. Diabetes Care 2010, 33, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Caprio, S.; Pierpont, B.; Van Name, M.; Savoye, M.; Parks, E.J. Hepatic De Novo Lipogenesis in Obese Youth Is Modulated by a Common Variant in the GCKR Gene. J. Clin. Endocrinol. Metab. 2015, 100, E1125–E1132. [Google Scholar] [CrossRef] [PubMed]
- Imamura, F.; Micha, R.; Khatibzadeh, S.; Fahimi, S.; Shi, P.; Powles, J.; Mozaffarian, D. Dietary quality among men and women in 187 countries in 1990 and 2010: A systematic assessment. Lancet Glob. Health 2015, 3, e132–e142. [Google Scholar] [CrossRef]
- Monteiro, C.A.; Moubarac, J.-C.; Cannon, G.; Ng, S.W.; Popkin, B. Ultra-processed products are becoming dominant in the global food system. Obes. Rev. 2013, 14, 21–28. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Bellanti, F.; Villani, R.; Facciorusso, A.; Vendemiale, G.; Serviddio, G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free. Radic. Biol. Med. 2017, 111, 173–185. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; MacIntyre, A.N.; Maciver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D. Dietary and Policy Priorities for Cardiovascular Disease, Diabetes, and Obesity: A Comprehensive Review. Circulation 2016, 133, 187–225. [Google Scholar] [CrossRef]
- Atkinson, F.S.; Foster-Powell, K.; Brand-Miller, J.C. International Tables of Glycemic Index and Glycemic Load Values: 2008. Diabetes Care 2008, 31, 2281–2283. [Google Scholar] [CrossRef]
- Hosseini, Z.; Whiting, S.J.; Vatanparast, H. Current evidence on the association of the metabolic syndrome and dietary patterns in a global perspective. Nutr. Res. Rev. 2016, 29, 152–162. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e5. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Mediterranean dietary pattern, inflammation and endothelial function: A systematic review and meta-analysis of intervention trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 929–939. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef]
- Ishiyama, J.; Taguchi, R.; Akasaka, Y.; Shibata, S.; Ito, M.; Nagasawa, M.; Murakami, K. Unsaturated FAs prevent palmitate-induced LOX-1 induction via inhibition of ER stress in macrophages. J. Lipid Res. 2011, 52, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.Q.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Gurav, A.; Sivaprakasam, S.; Bhutia, Y.D.; Boettger, T.; Singh, N.; Ganapathy, V. Slc5a8, a Na+-coupled high-affinity transporter for short-chain fatty acids, is a conditional tumour suppressor in colon that protects against colitis and colon cancer under low-fibre dietary conditions. Biochem. J. 2015, 469, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-Chain Fatty Acids Activate GPR41 and GPR43 on Intestinal Epithelial Cells to Promote Inflammatory Responses in Mice. Gastroenterology 2013, 145, 396–406.e10. [Google Scholar] [CrossRef]
- Nastasi, C.; Candela, M.; Bonefeld, C.M.; Geisler, C.; Hansen, M.; Krejsgaard, T.; Biagi, E.; Andersen, M.H.; Brigidi, P.; Ødum, N.; et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci. Rep. 2015, 5, 16148. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Papac-Milicevic, N.; Busch, C.-L.; Binder, C. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. Adv. Immunol. 2016, 131, 1–59. [Google Scholar] [CrossRef]
- Chou, M.-Y.; Fogelstrand, L.; Hartvigsen, K.; Hansen, L.F.; Woelkers, D.; Shaw, P.X.; Choi, J.; Perkmann, T.; Bäckhed, F.; Miller, Y.I.; et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Investig. 2009, 119, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Mottaran, E.; Vidali, M.; Reale, E.; Saksena, S.; Occhino, G.; Burt, A.D.; Day, C.P. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut 2005, 54, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.A.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, P.J.D.F.; Terabe, M.; Kapoor, V.; Elgindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nat. Cell Biol. 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; Van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.; Cyster, J.G. 25-Hydroxycholesterol suppresses interleukin-1–driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Blaak, E.E. Mitochondrial Dysfunction is a Key Pathway that Links Saturated Fat Intake to the Development and Progression of NAFLD. Mol. Nutr. Food Res. 2021, 65, e1900942. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Godos, J.; Loguercio, C.; Salomone, F.; Loguercio, C. Targeting gut-liver axis for the treatment of nonalcoholic steatohepatitis: Translational and clinical evidence. Transl. Res. 2016, 167, 116–124. [Google Scholar] [CrossRef]
- Kosteli, A.; Sugaru, E.; Haemmerle, G.; Martin, J.F.; Lei, J.; Zechner, R.; Ferrante, A.W. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J. Clin. Investig. 2010, 120, 3466–3479. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Nussbaum, J.C.; Liang, H.-E.; Van Dyken, S.J.; Cheng, L.E.; Mohapatra, A.; Chawla, A.; Locksley, R.M. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 2013, 210, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef]
- Kosaraju, R.; Guesdon, W.; Crouch, M.J.; Teague, H.L.; Sullivan, E.M.; Karlsson, E.A.; Schultz-Cherry, S.; Gowdy, K.; Bridges, L.C.; Reese, L.R.; et al. B Cell Activity Is Impaired in Human and Mouse Obesity and Is Responsive to an Essential Fatty Acid upon Murine Influenza Infection. J. Immunol. 2017, 198, 4738–4752. [Google Scholar] [CrossRef]
- Mulder, W.J.M.; Ochando, J.; Joosten, L.A.B.; Fayad, Z.A.; Netea, M.G. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 2019, 18, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Wing, R.R.; Bolin, P.; Brancati, F.L.; Bray, G.A.; Clark, J.M.; Coday, M.; Crow, R.S.; Curtis, J.M.; Egan, C.M.; Espeland, M.A.; et al. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N. Engl. J. Med. 2013, 369, 145–154. [Google Scholar] [CrossRef]
- Schmitz, J.; Evers, N.; Awazawa, M.; Nicholls, H.; Brönneke, H.; Dietrich, A.; Mauer, J.; Blüher, M.; Brüning, J. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol. Metab. 2016, 5, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S.; et al. Hyperglycemia Promotes Myelopoiesis and Impairs the Resolution of Atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Seijkens, T.; Hoeksema, M.A.; Beckers, L.; Smeets, E.; Meiler, S.; Levels, J.; Tjwa, M.; De Winther, M.P.J.; Lutgens, E. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 2014, 28, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, E.; Jaminon, A.; Van Berkel, T.J.C.; Van Eck, M. Diet-induced (epigenetic) changes in bone marrow augment atherosclerosis. J. Leukoc. Biol. 2014, 96, 833–841. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variants/SNPs/Protein Variants | Relative Effects/Association with | |
|---|---|---|---|
| Major and most common genetic determinants of MAFLD | PNPLA3 | rs738409 (I148M) | Disruption of triglycerides and phospholipids turnover and remodelling: increased hepatic fat accumulation; Disruption of retinol storage in HSCs leading to higher risk of inflammation, fibrosis, and HCC progression. |
| MBOAT 7 | rs641738 | Higher risk of MAFLD development, inflammation, fibrosis, and HCC progression. | |
| TM6SF2 | rs58542926 | Favouring liver fat accumulation;Protection against the development of cardiovascular diseases. | |
| Other genetic determinants involved in lipid metabolism | GCKR | rs1260326 | Increased de novo lipogenesis and worsened hepatic steatosis. |
| PPP1R3B | rs4841132 | Reduction of de novo lipogenesis and thus protection from hepatic fat accumulation. | |
| APOB | Several and different | Reduced VLDL export from hepatocytes. | |
| Other genetic determinants involved in oxidative stress imbalance | SOD2 | rs4880 | Higher oxidative stress and more advanced fibrosis. |
| UCP3 | rs1800849 | IR worsening, increased adiponectin levels, and NASH development. | |
| UCP2 | rs695366 | Higher insulin sensitivity and protection against liver damage. | |
| MARC1 | A165T | Lower hepatic fat accumulation and decreased levels of several biomarkers of liver disease. | |
| HFE | rs1800562 (C282Y) | Iron overload and related oxidative stress imbalance. | |
| Other genetic determinants involved in inflammation and fibrosis | TLR4 | D299G and T399I | Protection against fibrosis (in animal models). |
| IFNL4 | rs368234815 | Induction of severe inflammation. | |
| IFN/IL-28 | rs12979860 | Promotes inflammation and fibrosis (it is predictive for advanced stage of the disease). | |
| PCSK7 | rs236918 | Liver damage and altered fibrogenesis association. | |
| MERTK | rs4374383 | Protection against fibrosis. | |
| HSD17B13 | rs72613567 | Reduced risk of NASH (but not steatosis). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallio, M.; Romeo, M.; Gravina, A.G.; Masarone, M.; Larussa, T.; Abenavoli, L.; Persico, M.; Loguercio, C.; Federico, A. Nutrigenomics and Nutrigenetics in Metabolic- (Dysfunction) Associated Fatty Liver Disease: Novel Insights and Future Perspectives. Nutrients 2021, 13, 1679. https://doi.org/10.3390/nu13051679
Dallio M, Romeo M, Gravina AG, Masarone M, Larussa T, Abenavoli L, Persico M, Loguercio C, Federico A. Nutrigenomics and Nutrigenetics in Metabolic- (Dysfunction) Associated Fatty Liver Disease: Novel Insights and Future Perspectives. Nutrients. 2021; 13(5):1679. https://doi.org/10.3390/nu13051679
Chicago/Turabian StyleDallio, Marcello, Mario Romeo, Antonietta Gerarda Gravina, Mario Masarone, Tiziana Larussa, Ludovico Abenavoli, Marcello Persico, Carmelina Loguercio, and Alessandro Federico. 2021. "Nutrigenomics and Nutrigenetics in Metabolic- (Dysfunction) Associated Fatty Liver Disease: Novel Insights and Future Perspectives" Nutrients 13, no. 5: 1679. https://doi.org/10.3390/nu13051679
APA StyleDallio, M., Romeo, M., Gravina, A. G., Masarone, M., Larussa, T., Abenavoli, L., Persico, M., Loguercio, C., & Federico, A. (2021). Nutrigenomics and Nutrigenetics in Metabolic- (Dysfunction) Associated Fatty Liver Disease: Novel Insights and Future Perspectives. Nutrients, 13(5), 1679. https://doi.org/10.3390/nu13051679