The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM)


Abstract
1. Introduction
2. Common Rodent Models of MetS and T2DM
2.1. Mouse Models
2.1.1. C57BL Mouse Background
High-Fat Induced Diabetes in C57BL Mouse
Microbiome and Diet in the C57BL Mouse
2.1.2. Spiny Mouse
2.2. Common Rat Models
2.2.1. Spontaneously Diabetic Torii (SDT) Rat
2.2.2. Zucker Diabetic Fatty (ZDF) Rat
2.2.3. UCDavis-Type 2 Diabetes Mellitus (UCD-T2DM) Rat/ Zucker Diabetic Sprague Dawley (ZDSD or ZDSD/Pco) Rat
2.2.4. Goto-Kakizaki Rat
2.2.5. Koletsky Rat
2.3. Spontaneous T2DM in Rats
2.3.1. Nile Rat and Sand Rat
2.3.2. Sand Rat
3. The Nile Rat (Arvicanthis niloticus)
3.1. Nile Rat Background
3.1.1. Establishing Breeding Pairs
3.1.2. Separation of Pups
Growth Curve
3.1.3. Handling the Nile Rat
3.2. Diet-Induced T2DM Diabetes in the Nile Rat
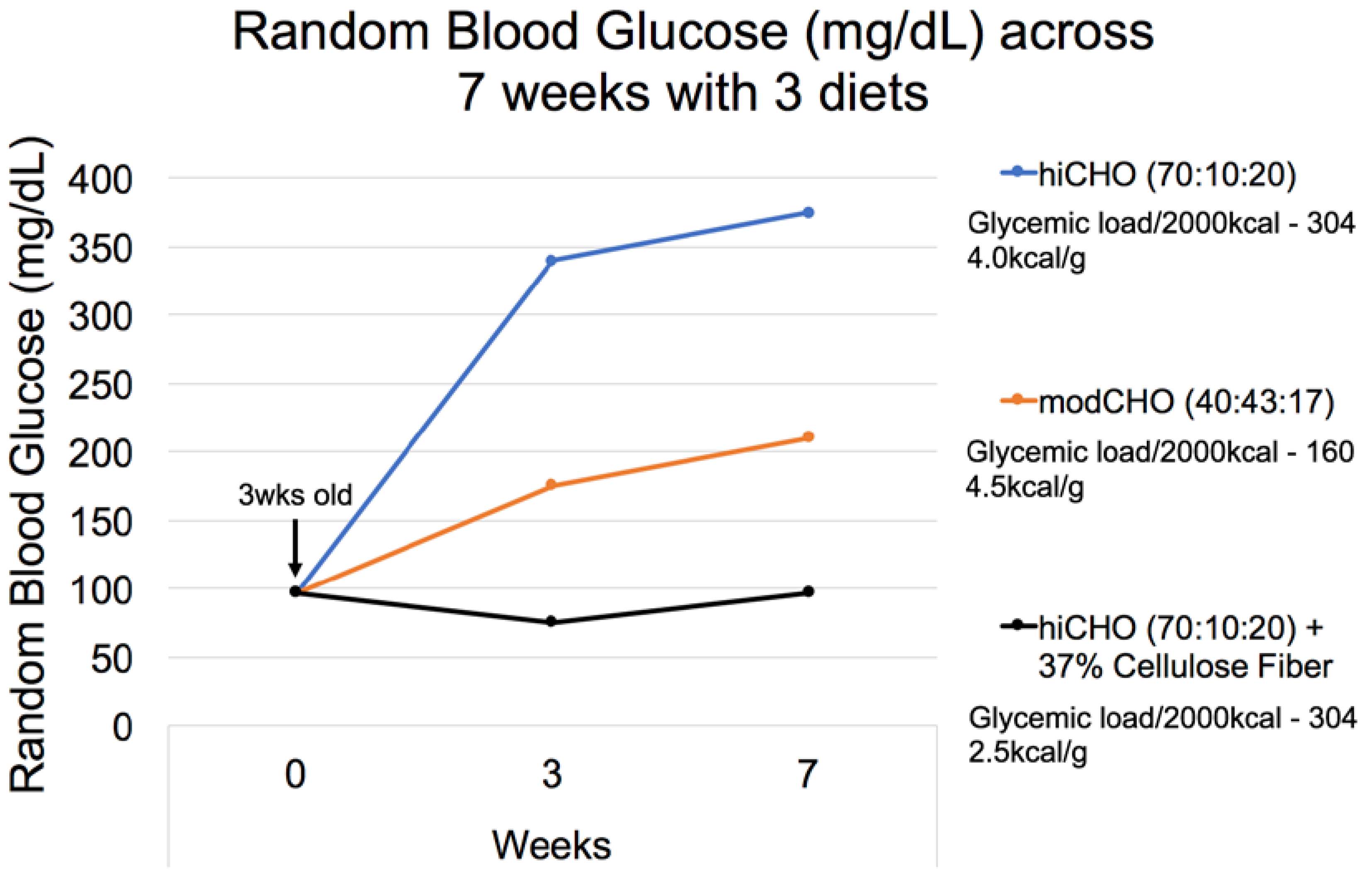
3.2.1. Diet Formulation
3.2.2. Dietary Factors and T2DM
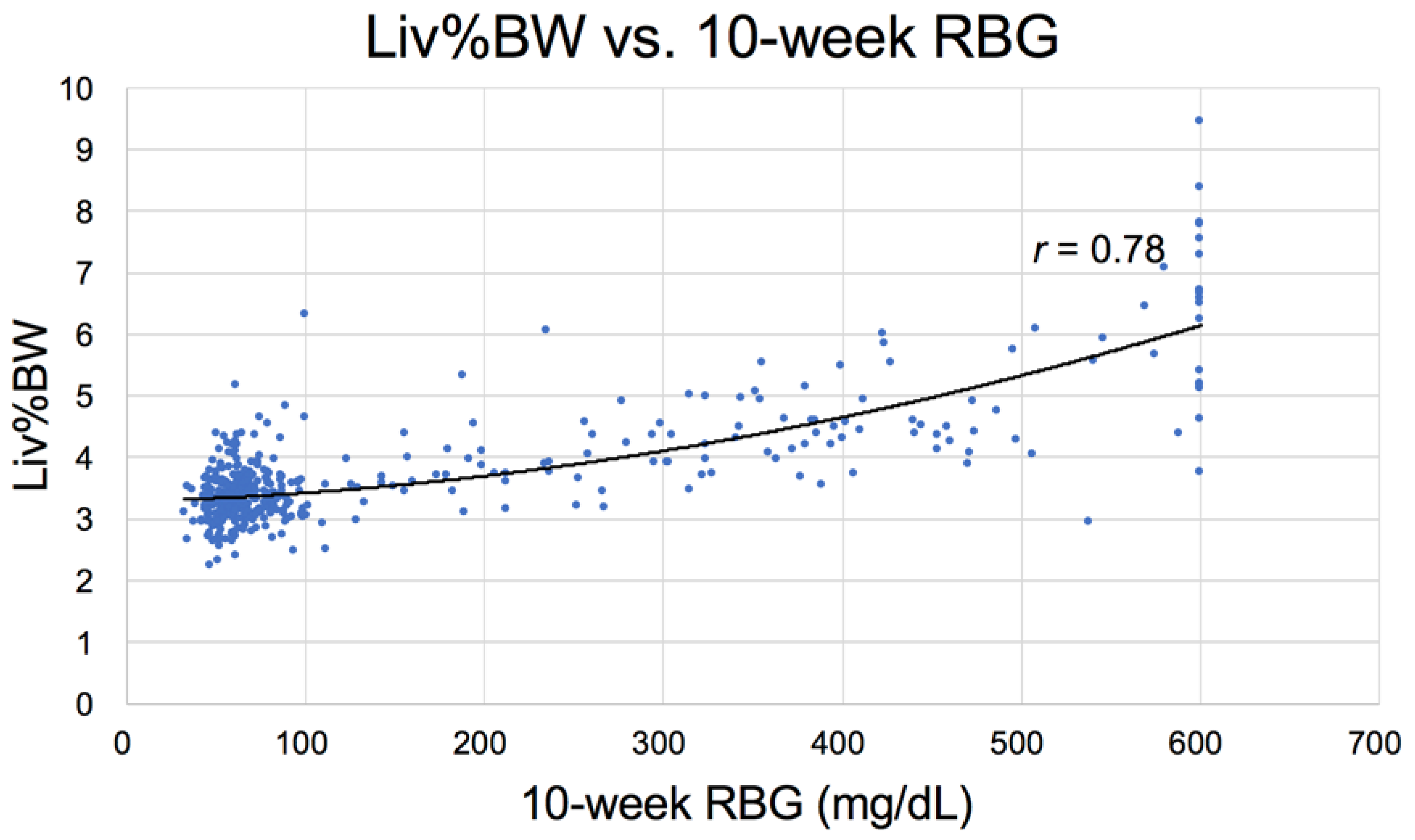
3.2.3. Physical Parameters
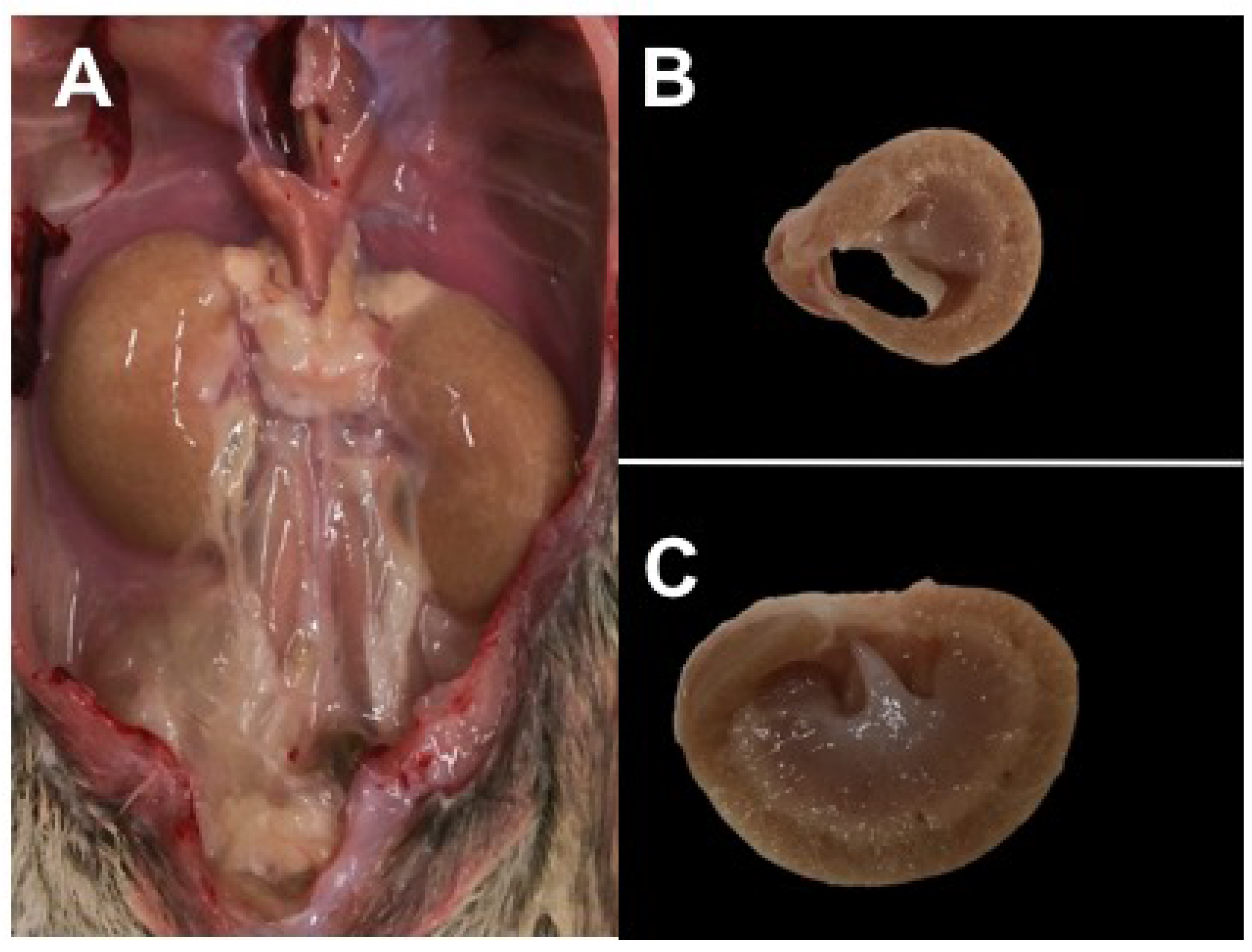
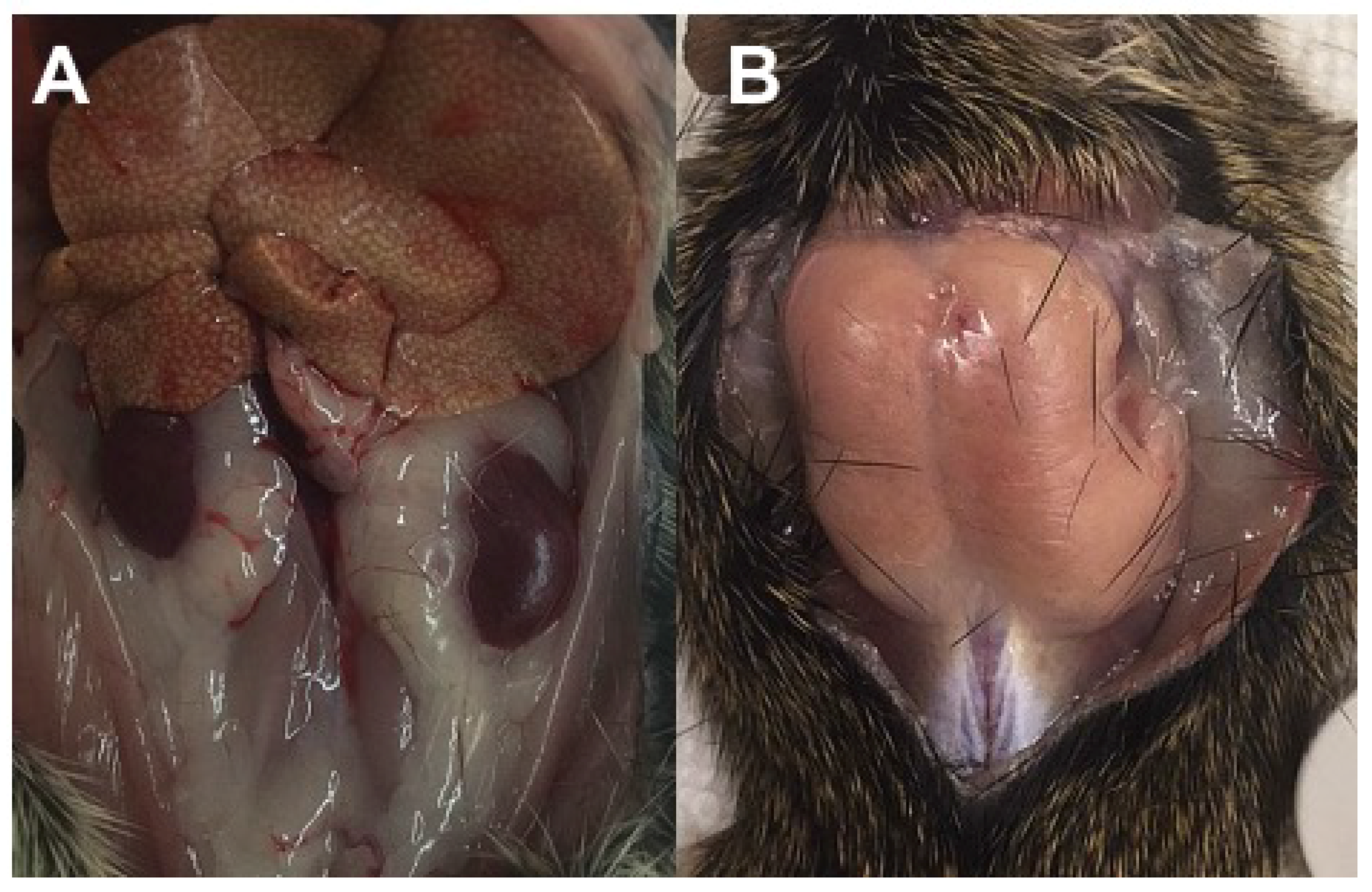
3.2.4. Diabetic Retinopathy
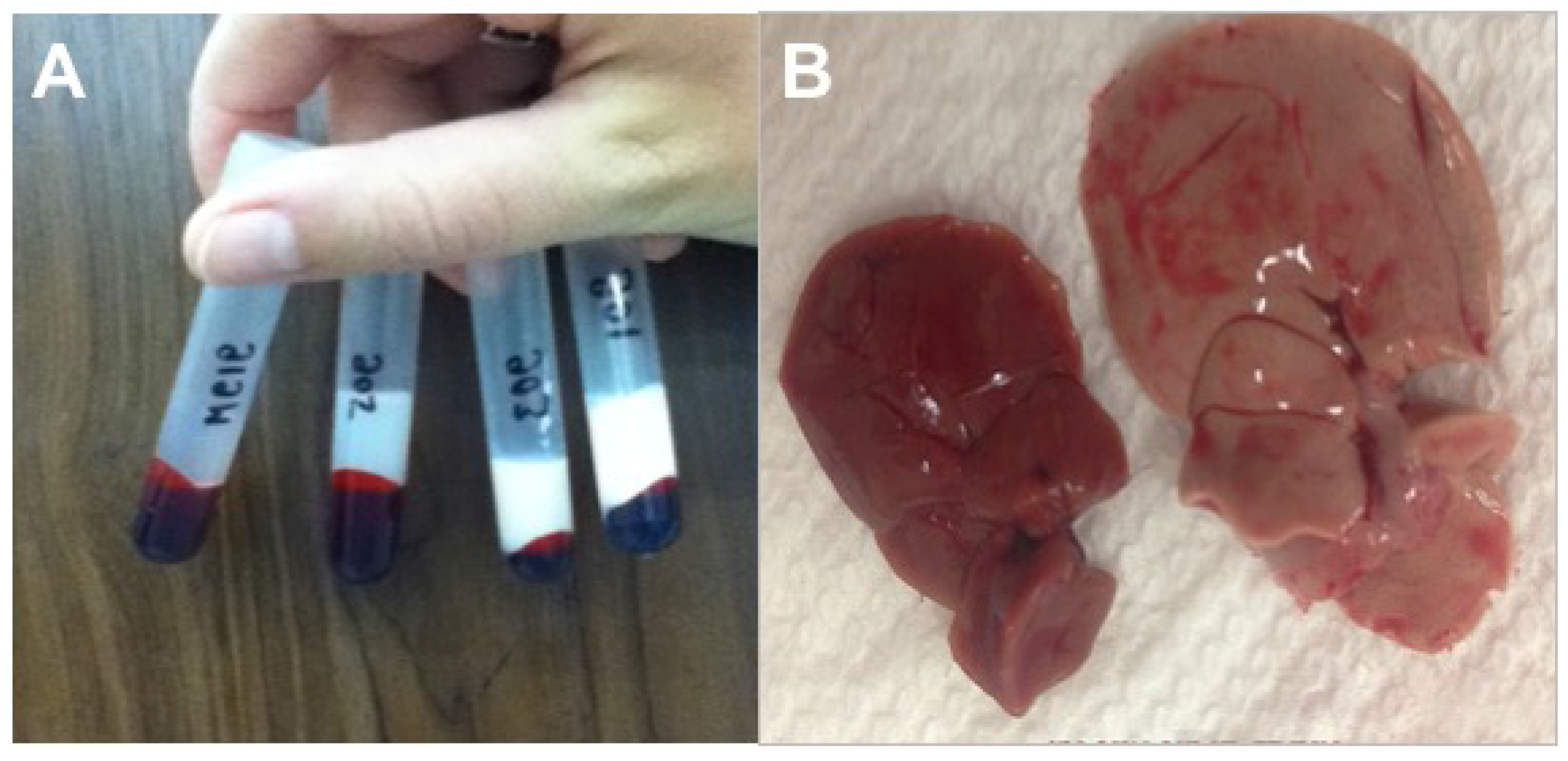
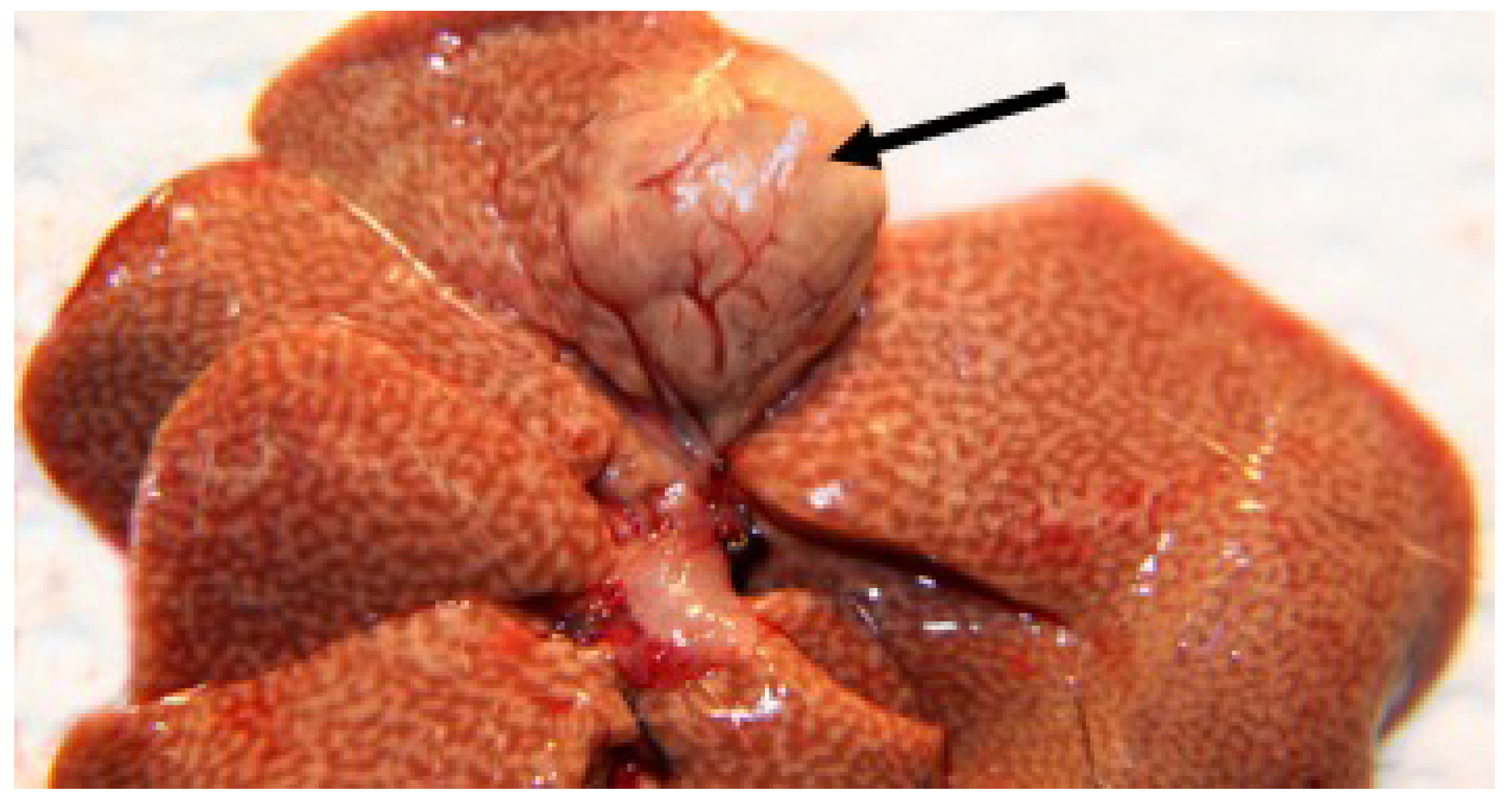
3.2.5. Necropsy Findings
3.2.6. Genetic Basis of Diabetes
3.2.7. Gender and Diabetes
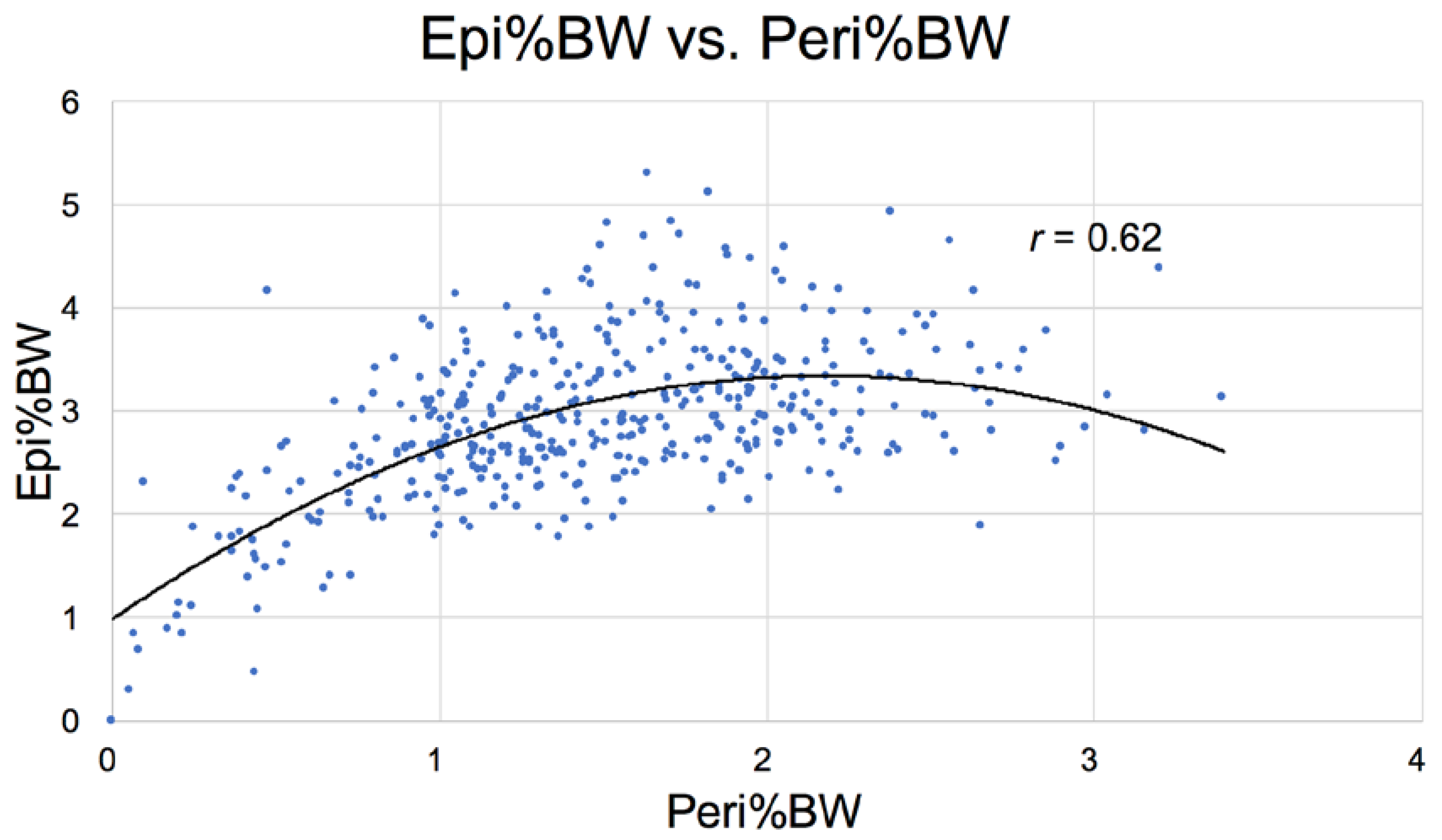
3.2.8. Food Intake and Adipose Pools
3.2.9. Dietary Fiber
3.2.10. Supplementation with Antidiabetic Agents and Polyphenols
4. Conclusions and Future Perspectives
Acknowledgments and Funding Support
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016; Volume 978, 88p, ISBN 978-92-4-156525-7. [Google Scholar]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. The Metabolic Syndrome—A New Worldwide Definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Reaven, G. The Metabolic Syndrome or the Insulin Resistance Syndrome? Different Names, Different Concepts, and Different Goals. Endocrinol. Metab. Clin. N. Am. 2004, 33, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-J.; Lee, H.-S.; Lee, J.-W. Association of Carbohydrate and Fat Intake with Metabolic Syndrome. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.; Imamura, F.; Brage, S.; Griffin, S.J.; Wareham, N.J.; Forouhi, N.G. Intakes and Sources of Dietary Sugars and Their Association with Metabolic and Inflammatory Markers. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Sharman, M.J.; Love, D.M.; Avery, N.G.; Gómez, A.L.; Scheett, T.P.; Kraemer, W.J. Body Composition and Hormonal Responses to a Carbohydrate-Restricted Diet. Metabolism 2002, 51, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Westman, E.C.; Yancy, W.S.; Olsen, M.K.; Dudley, T.; Guyton, J.R. Effect of a Low-Carbohydrate, Ketogenic Diet Program Compared to a Low-Fat Diet on Fasting Lipoprotein Subclasses. Int. J. Cardiol. 2006, 110, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The Effect of a Low-Carbohydrate, Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. Dig. Dis. Sci. 2007, 52, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary Carbohydrate Restriction as the First Approach in Diabetes Management: Critical Review and Evidence Base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, C.E.; Phinney, S.D.; Fernandez, M.L.; Quann, E.E.; Wood, R.J.; Bibus, D.M.; Kraemer, W.J.; Feinman, R.D.; Volek, J.S. Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2008, 43, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.O.; DeOgburn, R.; Ratliff, J.C.; Su, R.; Volek, J.S.; McGrane, M.M.; Dardik, A.; Fernandez, M.L. Low-Carbohydrate Diet Disrupts the Association between Insulin Resistance and Weight Gain. Metabolism 2009, 58, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of Low-Calorie versus Low-Carbohydrate Ketogenic Diet in Type 2 Diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Lam, Y.Y.; Leibel, R.L.; Mayer, L.E.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; et al. Energy Expenditure and Body Composition Changes after an Isocaloric Ketogenic Diet in Overweight and Obese Men. Am. J. Clin. Nutr. 2016, 104, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Noakes, T.; Volek, J.S.; Phinney, S.D. Low-Carbohydrate Diets for Athletes: What Evidence? Br. J. Sports Med. 2014, 48, 1077–1078. [Google Scholar] [CrossRef] [PubMed]
- Herman, W.H.; Zimmet, P. Type 2 Diabetes: An Epidemic Requiring Global Attention and Urgent Action. Diabetes Care 2012, 35, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Editorial. Beat Diabetes: An Urgent Call for Global Action. Lancet 2016, 387, 1483. [Google Scholar]
- Krug, E.G. Trends in Diabetes: Sounding the Alarm. Lancet 2016, 387, 1485–1486. [Google Scholar] [CrossRef]
- Ezzati, M. Worldwide Trends in Diabetes since 1980: A Pooled Analysis of 751 Population-Based Studies with 4·4 Million Participants. Lancet 2016, 387, 1513–1530. [Google Scholar]
- Pratley, R.E. Gene-Environment Interactions in the Pathogenesis of Type 2 Diabetes Mellitus: Lessons Learned from the Pima Indians. Proc. Nutr. Soc. 1998, 57, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Dorcely, B.; Katz, K.; Jagannathan, R.; Chiang, S.S.; Oluwadare, B.; Goldberg, I.J.; Bergman, M. Novel Biomarkers for Prediabetes, Diabetes, and Associated Complications. Diabetes Metab. Syndr. Obes. 2017, 10, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B. Globalization of Diabetes. Diabetes Care 2011, 34, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Chin, K.-Y.; Suhaimi, F.H.; Fairus, A.; Ima-Nirwana, S. Animal Models of Metabolic Syndrome: A Review. Nutr. Metab. 2016, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.R.; Wagner, J.D. Type 2 Diabetes-an Introduction to the Development and Use of Animal Models. ILAR J. 2006, 47, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, G.; Yuli, M.; Donath, M.Y.; Nesher, R.; Melloul, D.; Cerasi, E.; Gross, D.J.; Kaiser, N. Beta-Cell Glucotoxicity in the Psammomys obesus Model of Type 2 Diabetes. Diabetes 2001, 50 (Suppl. 1), S113–S117. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.P.; Digitale, E.K.; Stanhope, K.L.; Graham, J.L.; Baskin, D.G.; Reed, B.J.; Sweet, I.R.; Griffen, S.C.; Havel, P.J. Development and Characterization of a Novel Rat Model of Type 2 Diabetes Mellitus: The UC Davis Type 2 Diabetes Mellitus UCD-T2DM Rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1782–R1793. [Google Scholar] [CrossRef] [PubMed]
- Reinwald, S.; Peterson, R.G.; Allen, M.R.; Burr, D.B. Skeletal Changes Associated with the Onset of Type 2 Diabetes in the ZDF and ZDSD Rodent Models. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E765–E774. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, R.J.; Karim, L.; Calley, V.I.; Bouxsein, M.L. A Review of Rodent Models of Type 2 Diabetic Skeletal Fragility. J. Bone Miner. Res. 2014, 29, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Leow, S.-S.; Bolsinger, J.; Pronczuk, A.; Hayes, K.C.; Sambanthamurthi, R. Hepatic Transcriptome Implications for Palm Fruit Juice Deterrence of Type 2 Diabetes Mellitus in Young Male Nile Rats. Genes Nutr. 2016, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Beddow, S.A.; Iwasaki, T.; Zhang, X.-M.; Chu, X.; Still, C.D.; Gerhard, G.S.; Shulman, G.I. Fasting Hyperglycemia Is Not Associated with Increased Expression of PEPCK or G6Pc in Patients with Type 2 Diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 12121–12126. [Google Scholar] [CrossRef] [PubMed]
- Bolsinger, J.; Landstrom, M.; Pronczuk, A.; Auerbach, A.; Hayes, K.C. Low Glycemic Load Diets Protect against Metabolic Syndrome and Type 2 Diabetes Mellitus in the Male Nile Rat. J. Nutr. Biochem. 2017, 42, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Ernsberger, P.; Koletsky, R.J.; Friedman, J.E. Molecular Pathology in the Obese Spontaneous Hypertensive Koletsky Rat: A Model of Syndrome X. Ann. N. Y. Acad. Sci. 1999, 892, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Aleixandre de Artiñano, A.; Miguel Castro, M. Experimental Rat Models to Study the Metabolic Syndrome. Br. J. Nutr. 2009, 102, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Chaabo, F.; Pronczuk, A.; Maslova, E.; Hayes, K. Nutritional Correlates and Dynamics of Diabetes in the Nile Rat (Arvicanthis niloticus): A Novel Model for Diet-Induced Type 2 Diabetes and the Metabolic Syndrome. Nutr. Metab. 2010, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Melhorn, M.I.; Zandi, S.; Frimmel, S.; Tayyari, F.; Hisatomi, T.; Almulki, L.; Pronczuk, A.; Hayes, K.C.; Hafezi-Moghadam, A. An Animal Model of Spontaneous Metabolic Syndrome: Nile Grass Rat. FASEB J. 2010, 24, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Bolsinger, J.; Pronczuk, A.; Hayes, K.C. Dietary Carbohydrate Dictates Development of Type 2 Diabetes in the Nile Rat. J. Nutr. Biochem. 2013, 24, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated Death of Retinal Microvascular Cells in Human and Experimental Diabetic Retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Sambanthamurthi, R.; Tan, Y.; Sundram, K.; Abeywardena, M.; Sambandan, T.G.; Rha, C.; Sinskey, A.J.; Subramaniam, K.; Leow, S.-S.; Hayes, K.C.; et al. Oil Palm Vegetation Liquor: A New Source of Phenolic Bioactives. Br. J. Nutr. 2011, 106, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Gotzmann, J.; Kuny, S.; Huang, H.; Sauve, Y.; Chan, C.B. Five Stages of Progressive Beta-Cell Dysfunction in the Laboratory Nile Rat Model of Type 2 Diabetes. J. Endocrinol. 2016, 229, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Shafrir, E.; Ziv, E.; Kalman, R. Nutritionally Induced Diabetes in Desert Rodents as Models of Type 2 Diabetes: Acomys cahirinus (Spiny Mice) and Psammomys obesus (Desert Gerbil). ILAR J. 2006, 47, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Kanety, H.; Moshe, S.; Shafrir, E.; Lunenfeld, B.; Karasik, A. Hyperinsulinemia Induces a Reversible Impairment in Insulin Receptor Function Leading to Diabetes in the Sand Rat Model of Non-Insulin-Dependent Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 1994, 91, 1853–1857. [Google Scholar] [CrossRef] [PubMed]
- Walder, K.; Oakes, N.; Fahey, R.P.; Cooney, G.; Zimmet, P.Z.; Collier, G.R. Profile of Dyslipidemia in Psammomys obesus, an Animal Model of the Metabolic Syndrome. Endocr. Regul. 2002, 36, 1–8. [Google Scholar] [PubMed]
- Walder, K.R.; Fahey, R.P.; Morton, G.J.; Zimmet, P.Z.; Collier, G.R. Characterization of Obesity Phenotypes in Psammomys obesus (Israeli Sand rats). Int. J. Exp. Diabetes Res. 2000, 1, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hackel, D.B.; Lebovitz, H.E.; Frohman, L.A.; Mikat, E.; Schmidt-Nielsen, K. Effect of Caloric Restriction on the Glucose Tolerance and Plasma Insulin of the Sand rat. Metabolism 1967, 16, 1133–1139. [Google Scholar] [CrossRef]
- Kalman, R.; Adler, J.H.; Lazarovici, G.; Bar-On, H.; Ziv, E. The Efficiency of Sand rat Metabolism Is Responsible for Development of Obesity and Diabetes. J. Basic Clin. Physiol. Pharmacol. 1993, 4, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Ziv, E.; Shafrir, E.; Kalman, R.; Galer, S.; Bar-On, H. Changing Pattern of Prevalence of Insulin Resistance in Psammomys obesus, a Model of Nutritionally Induced Type 2 Diabetes. Metabolism 1999, 48, 1549–1554. [Google Scholar] [CrossRef]
- Kaiser, N.; Nesher, R.; Donath, M.Y.; Fraenkel, M.; Behar, V.; Magnan, C.; Ktorza, A.; Cerasi, E.; Leibowitz, G. Psammomys obesus, a Model for Environment-Gene Interactions in Type 2 Diabetes. Diabetes 2005, 54 (Suppl. 2), S137–S144. [Google Scholar] [CrossRef] [PubMed]
- Hillel, J.; Gefel, D.; Kalman, R.; Ben-Ari, G.; David, L.; Orion, O.; Feldman, M.W.; Bar-On, H.; Blum, S.; Raz, I.; et al. Evidence for a Major Gene Affecting the Transition from Normoglycaemia to Hyperglycaemia in Psammomys obesus. Heredity 2005, 95, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Kalman, R.; Ziv, E.; Shafrir, E.; Bar-On, H.; Perez, R. Psammomys obesus and the Albino Rat--Two Different Models of Nutritional Insulin Resistance, Representing Two Different Types of Human Populations. Lab. Anim. 2001, 35, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.D.; Zhang, N.N.; Sokoloff, G.; Fanselow, M.S.; Ennes, H.S.; Palmer, A.A.; McRoberts, J.A. Behavioral Differences among C57BL/6 Substrains: Implications for Transgenic and Knockout Studies. J. Neurogenet. 2008, 22, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Morse, H. Origins of Inbred Mice; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Mekada, K.; Abe, K.; Murakami, A.; Nakamura, S.; Nakata, H.; Moriwaki, K.; Obata, Y.; Yoshiki, A.; Yoshiki, A. Genetic Differences among C57BL/6 Substrains. Exp. Anim. 2009, 58, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.Z.; Roussos, E.T.; Péterfy, M. Genetic Analysis of the Diabetes-Prone C57BLKS/J Mouse Strain Reveals Genetic Contribution from Multiple Strains. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Martin, T.L.; Surwit, R.S.; Robidoux, J. Genetic Vulnerability to Diet-Induced Obesity in the C57BL/6J Mouse: Physiological and Molecular Characteristics. Physiol. Behav. 2004, 81, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital Leptin Deficiency Is Associated with Severe Early-Onset Obesity in Humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Sun, G.-D.; Sun, J.; Liu, S.-J.; Wang, J.; Xu, X.-H.; Miao, L.-N. Spontaneous Type 2 Diabetic Rodent Models. J. Diabetes Res. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Borghjid, S.; Feinman, R. Response of C57Bl/6 Mice to a Carbohydrate-Free Diet. Nutr. Metab. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffé-Scrive, M. Differential Effects of Fat and Sucrose on the Development of Obesity and Diabetes in C57BL/6J and A/J Mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]
- Schreyer, S.A.; Wilson, D.L.; Leboeuf, R.C. C57BL/6 Mice Fed High Fat Diets as Models for Diabetes-Accelerated Atherosclerosis. Atherosclerosis 1998, 136, 17–24. [Google Scholar] [CrossRef]
- Guo, J.; Jou, W.; Gavrilova, O.; Hall, K.D. Persistent Diet-Induced Obesity in Male C57BL/6 Mice Resulting from Temporary Obesigenic Diets. PLoS ONE 2009, 4, e5370. [Google Scholar] [CrossRef] [PubMed]
- West, D.B.; Boozer, C.N.; Moody, D.L.; Atkinson, R.L. Dietary Obesity in Nine Inbred Mouse Strains. Am. J. Physiol. 1992, 262, R1025–R1032. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity Alters Gut Microbial Ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Diamant, M.; Blaak, E.E.; de Vos, W.M. Do Nutrient-Gut-Microbiota Interactions Play a Role in Human Obesity, Insulin Resistance and Type 2 Diabetes? Obes. Rev. 2011, 12, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Pellizzon, M.A.; Ricci, M.R. The Common Use of Improper Control Diets in Diet-Induced Metabolic Disease Research Confounds Data Interpretation: The Fiber Factor. Nutr. Metab. 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Suzuki, K.I.; Ono, T.; Sasaki, M.; Toyota, T. Development of Diabetes in the Non-Obese NIDDM Rat (GK Rat). Prediabetes Adv. Exp. Med. Biol. 1988, 246, 29–31. [Google Scholar] [PubMed]
- Akash, M.S.; Rehman, K.; Chen, S. Goto-Kakizaki Rats: Its Suitability as Non-Obese Diabetic Animal Model for Spontaneous Type 2 Diabetes Mellitus. Curr. Diabetes Rev. 2013, 9, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dubois, D.C.; Cao, Y.; Jusko, W.J.; Almon, R.R. Diabetes Disease Progression in Goto-Kakizaki Rats: Effects of Salsalate Treatment. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 381–389. [Google Scholar]
- Maekawa, F.; Fujiwara, K.; Kohno, D.; Kuramochi, M.; Kurita, H.; Yada, T. Young Adult-Specific Hyperphagia in Diabetic Goto-Kakizaki Rats Is Associated with Leptin Resistance and Elevation of Neuropeptide Y mRNA in the Arcuate Nucleus. J. Neuroendocrinol. 2006, 18, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Beddow, S.A.; Samuel, V.T. Fasting Hyperglycemia in the Goto-Kakizaki Rat Is Dependent on Corticosterone: A Confounding Variable in Rodent Models of Type 2 Diabetes. Dis. Model. Mech. 2012, 5, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, T.; Fuse, M.; Yokoi, N.; Shinohara, M.; Tsujii, H.; Kanazawa, M.; Kanazawa, Y.; Komeda, K.; Taniguchi, K. Genetic Analysis for Diabetes in a New Rat Model of Nonobese Type 2 Diabetes, Spontaneously Diabetic Torii Rat. Biochem. Biophys. Res. Commun. 2003, 304, 196–206. [Google Scholar] [CrossRef]
- Masuyama, T.; Komeda, K.; Hara, A.; Noda, M.; Shinohara, M.; Oikawa, T.; Kanazawa, Y.; Taniguchi, K. Chronological Characterization of Diabetes Development in Male Spontaneously Diabetic Torii Rats. Biochem. Biophys. Res. Commun. 2004, 314, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, H.; Yamamoto, H.; Sakata, K.; Fukuda, S.; Ito, M.; Sasase, T.; Miyajima, K.; Ueda, N.; Ohta, T.; Matsushita, M. Characterization of Hepatic Glucose Metabolism Disorder with the Progress of Diabetes in Male Spontaneously Diabetic Torii Rats. J. Vet. Med. Sci. 2008, 70, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Ohta, T.; Sasase, T.; Morinaga, H.; Hata, T.; Miyajima, K.; Katusda, Y.; Masuyama, T.; Shinohara, M.; Kakutani, M.; et al. A High-Fat Diet Inhibits the Progression of Diabetes Mellitus in Type 2 Diabetic Rats. Nutr. Res. 2010, 30, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N. Genetics of the Spontaneously Diabetic Torii Rat. Open Diabetes J. 2011, 4, 21–25. [Google Scholar] [CrossRef]
- Fuse, M.; Yokoi, N.; Shinohara, M.; Masuyama, T.; Kitazawa, R.; Kitazawa, S.; Seino, S. Identification of a Major Locus for Islet Inflammation and Fibrosis in the Spontaneously Diabetic Torii Rat. Physiol. Genom. 2008, 35, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Masuyama, T.; Shoda, T.; Takahashi, T.; Katsuda, Y.; Komeda, K.; Kuroki, M.; Kakehashi, A.; Kanazaw, Y. A New Spontaneously Diabetic Non-Obese Torii Rat Strain with Severe Ocular Complications. Int. J. Exp. Diabetes Res. 2000, 1, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, T.; Katsuda, Y.; Shinohara, M. A Novel Model of Obesity-Related Diabetes: Introgression of the Lepr(fa) Allele of the Zucker Fatty Rat into Nonobese Spontaneously Diabetic Torii (SDT) Rats. Exp. Anim. 2005, 54, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, J.P.; Sparks, J.D.; Peterson, R.G.; Smith, R.L.; Sparks, C.E. Effect of Dietary Fat on the Development of Non-Insulin Dependent Diabetes Mellitus in Obese Zucker Diabetic Fatty Male and Female Rats. Atherosclerosis 2000, 148, 231–241. [Google Scholar] [CrossRef]
- Davidson, E.P.; Coppey, L.J.; Holmes, A.; Lupachyk, S.; Dake, B.L.; Oltman, C.L.; Peterson, R.G.; Yorek, M.A. Characterization of Diabetic Neuropathy in the Zucker Diabetic Sprague-Dawley Rat: A New Animal Model for Type 2 Diabetes. J. Diabetes Res. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.G.; Jackson, C.V.; Zimmerman, K.; De Winter, W.; Huebert, N.; Hansen, M.K. Characterization of the ZDSD Rat: A Translational Model for the Study of Metabolic Syndrome and Type 2 Diabetes. J. Diabetes Res. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.E.; Cain, J.; Banz, W.J.; Peterson, R.G. Age-Related Differences in Response to High-Fat Feeding on Adipose Tissue and Metabolic Profile in ZDSD Rats. ISRN Obes. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Livesey, G.; Taylor, R.; Hulshof, T.; Howlett, J. Glycemic Response and Health—A Systematic Review and Meta-Analysis: Relations between Dietary Glycemic Properties and Health Outcomes. Am. J. Clin. Nutr. 2008, 87, 258S–268S. [Google Scholar] [CrossRef] [PubMed]
- Wolever, T.M. Effect of Macronutrients on the Glycemic Index. Am. J. Clin. Nutr. 2017, 3, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Krajmalnik-Brown, R.; Ilhan, Z.-E.; Kang, D.-W.; DiBaise, J.K. Effects of Gut Microbes on Nutrient Absorption and Energy Regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Cani, P.D. Diabetes, Obesity and Gut Microbiota. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Shafrir, E. Overnutrition in Spiny Mice (Acomys cahirinus): β-Cell Expansion Leading to Rupture and Overt Diabetes on Fat-Rich Diet and Protective Energy-Wasting Elevation in Thyroid Hormone on Sucrose-Rich Diet. Diabetes. Metab. Res. Rev. 2000, 16, 94–105. [Google Scholar] [CrossRef]
- Gonzalez, A.D.; Gallant, M.A.; Burr, D.B.; Wallace, J.M. Multiscale Analysis of Morphology and Mechanics in Tail Tendon from the ZDSD Rat Model of Type 2 Diabetes. J. Biomech. 2014, 47, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.P.; Stanhope, K.L.; Graham, J.L.; Evans, J.L.; Baskin, D.G.; Griffen, S.C.; Havel, P.J. Dietary Fructose Accelerates the Development of Diabetes in UCD-T2DM Rats: Amelioration by the Antioxidant, α-Lipoic Acid. AJP Regul. Integr. Comp. Physiol. 2010, 298, R1343–R1350. [Google Scholar] [CrossRef] [PubMed]
- Degen, A.A.; Kam, M.; Khokhlova, I.S.; Zeevi, Y. Fiber Digestion and Energy Utilization of Fat Sand rats (Psammomys obesus) Consuming the Chenopod Anabasis Articulata. Physiol. Biochem. Zool. 2000, 73, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Faris, M.A.I.E.; Takruri, H.R.; Issa, A.Y. Role of Lentils (Lens Culinaris L.) in Human Health and Nutrition: A Review. Med. J. Nutr. Metab. 2013, 6, 3–16. [Google Scholar] [CrossRef]
- Wegh, C.A.M.; Schoterman, M.H.C.; Vaughan, E.E.; Belzer, C.; Benninga, M.A. The Effect of Fiber and Prebiotics on Children’s Gastrointestinal Disorders and Microbiome. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Cantero, I.; Abete, I.; Monreal, J.I.; Martinez, J.A.; Zulet, M.A. Fruit Fiber Consumption Specifically Improves Liver Health Status in Obese Subjects under Energy Restriction. Nutrients 2017, 9, 667. [Google Scholar] [CrossRef] [PubMed]
- Lattimer, J.M.; Haub, M.D. Effects of Dietary Fiber and Its Components on Metabolic Health. Nutrients 2010, 2, 1266–1289. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarczyk, M.M.; Miller, M.J.; Freund, G.G. The Health Benefits of Dietary Fiber: Beyond the Usual Suspects of Type 2 Diabetes Mellitus, Cardiovascular Disease and Colon Cancer. Metabolism 2012, 61, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between Host Genetics and Gut Microbiome in Diabetes and Metabolic Syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Nakao, S.; Zandi, S.; Sun, D.; Hayes, K.C.; Hafezi-Moghadam, A. Retinopathy in a Novel Model of Metabolic Syndrome and Type 2 Diabetes: New Insight on the Inflammatory Paradigm. FASEB J. 2014, 28, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Han, W.H.; Gotzmann, J.; Kuny, S.; Huang, H.; Chan, C.B.; Lemieux, H.; Sauvé, Y. Modifications in Retinal Mitochondrial Respiration Precede Type 2 Diabetes and Protracted Microvascular Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3826–3839. [Google Scholar] [CrossRef] [PubMed]
- Esparza-Romero, J.; Valencia, M.E.; Urquidez-Romero, R.; Chaudhari, L.S.; Hanson, R.L.; Knowler, W.C.; Ravussin, E.; Bennett, P.H.; Schulz, L.O. Environmentally Driven Increases in Type 2 Diabetes and Obesity in Pima Indians and Non-Pimas in Mexico Over a 15-Year Period: The Maycoba Project. Diabetes Care 2015, 38, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.O.; Chaudhari, L.S. High-Risk Populations: The Pimas of Arizona and Mexico. Curr. Obes. Rep. 2015, 4, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.; Brown, F.; Remillard, D.E.; Bolsinger, J.; Hayes, K.C. Pathology of the Nile Rat Developing Type 2 Diabetes. FASEB J. 2013, 27. [Google Scholar]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-Mediated Dysbiosis Regulates Progression of NAFLD and Obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Wang, N.; Qin, W. Gut Microbiota and Hepatocellular Carcinoma. Gastrointest. Tumors 2015, 2, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Darnaud, M.; Faivre, J.; Moniaux, N. Targeting Gut Flora to Prevent Progression of Hepatocellular Carcinoma. J. Hepatol. 2013, 58, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients 2017, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, A.D.; Zhou, L.; Christensen, J.; Marlétaz, F.; Liu, S.; Li, F.; Jansen, P.G.; Spiga, E.; Hansen, M.T.; Pedersen, S.V.H.; et al. Genome Sequence of a Diabetes-Prone Rodent Reveals a Mutation Hotspot around the ParaHox Gene Cluster. Proc. Natl. Acad. Sci. USA 2017, 114, 7677–7682. [Google Scholar] [CrossRef] [PubMed]
- Frankenfeld, C.L.; Atkinson, C.; Wähälä, K.; Lampe, J.W. Obesity Prevalence in Relation to Gut Microbial Environments Capable of Producing Equol or O-Desmethylangolensin from the Isoflavone Daidzein. Eur. J. Clin. Nutr. 2014, 68, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Frankenfeld, C.L. Cardiometabolic Risk and Gut Microbial Phytoestrogen Metabolite Phenotypes. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.M.; Lampe, J.W.; Newton, K.M.; Gundersen, G.; Fuller, S.; Reed, S.D.; Frankenfeld, C.L. Being Overweight or Obese Is Associated with Harboring a Gut Microbial Community Not Capable of Metabolizing the Soy Isoflavone Daidzein to O-Desmethylangolensin in Peri- and Post-Menopausal Women. Maturitas 2017, 99, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Vosselman, M.J.; van Marken Lichtenbelt, W.D.; Schrauwen, P. Energy Dissipation in Brown Adipose Tissue: From Mice to Men. Mol. Cell. Endocrinol. 2013, 379, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Vosselman, M.J.; Brans, B.; Van Der Lans, A.A.J.J.; Wierts, R.; Van Baak, M.A.; Mottaghy, F.M.; Schrauwen, P.; Van Marken Lichtenbelt, W.D. Brown Adipose Tissue Activity after a High-Calorie Meal in Humans. Am. J. Clin. Nutr. 2013, 98, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Chondronikola, M.; Volpi, E.; Børsheim, E.; Porter, C.; Annamalai, P.; Enerbäck, S.; Lidell, M.E.; Saraf, M.K.; Labbe, S.M.; Hurren, N.M.; et al. Brown Adipose Tissue Improves Whole-Body Glucose Homeostasis and Insulin Sensitivity in Humans. Diabetes 2014, 63, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Chondronikola, M.; Volpi, E.; Børsheim, E.; Porter, C.; Saraf, M.K.; Annamalai, P.; Yfanti, C.; Chao, T.; Wong, D.; Shinoda, K.; et al. Brown Adipose Tissue Activation Is Linked to Distinct Systemic Effects on Lipid Metabolism in Humans. Cell Metab. 2016, 23, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; DeFronzo, R.A.; Rosenstock, J.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results from Short-Term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care 2016, 39, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Sambanthamurthi, R.; Tan, Y.; Sundram, K.; Hayes, K.C.; Abeywardena, M.; Leow, S.-S.; Devi Sekaran, S.; Sambandan, T.G.; Rha, C.; Sinskey, A.J.; et al. Positive Outcomes of Oil Palm Phenolics on Degenerative Diseases in Animal Models. Br. J. Nutr. 2011, 106, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Bolsinger, J.; Pronczuk, A.; Sambanthamurthi, R.; Hayes, K.C. Anti-Diabetic Effects of Palm Fruit Juice in the Nile Rat (Arvicanthis niloticus). J. Nutr. Sci. 2014, 3, e5. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Development of MetS | Diabetes Induced by | Signs of Metabolic Syndrome | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hyperphagia with Cause | Incr BP * | Abdominal Obesity | Incr TG * | Decr HDL * | Hyper Insulinemia | |||||
| Human [2,3,4,5,20,21,29] | Natural (diet x gene interaction) | Carbohydrate | No | Diabetics, hiGLoad * diets | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Nile rat [28,30,31,32,33,34,35,36,37,38] | Natural (diet x gene interaction) | Carbohydrate | No | Diabetics, hiGLoad diets | ✓ | no | ✓ | ✓ | ✓ | |
| Sand rat [23,24,39,40,41,42,43,44,45,46,47,48] | Selective Breeding | (diet x gene interaction) | Carbohydrate | No | Diabetics fed chow | No | ✓ | ✓ | ✓ | ✓ |
| C57BL/6J mouse [22,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66] | Spontaneous Mutation | Lep(ob);lep deficient | Fat | No | Increased feed efficiency | ✓ | ✓ | No | No | ✓ |
| Goto-Kakizaki rat [22,67,68,69,70,71,72] | Spontaneous Mutation | NPY mRNA excess | Carbohydrate | ✓ | Leptin Resistance | No | No | No | N/A | ✓ |
| SDT rat * [73,74,75,76,77,78,79,80] | Spontaneous Mutation | Lepr(fa) | Carbohydrate | ✓ | Mutated Leptin Receptor | N/A | No | ✓ | N/A | ✓ |
| Koletsky rat [31] | Spontaneous Mutation | fa(k) | Carbohydrate | ✓ | Mutated Leptin Receptor | ✓ | ✓ | ✓ | N/A | ✓ |
| ZDF rat * [22,26,81] | Spontaneous Mutation | fa/fa | Fat | ✓ | Mutated Leptin Receptor | ✓ | ✓ | ✓ | ✓ | ✓ |
| UCD-T2DM rat/ZDSD rat * [25,26,27,82,83,84] | Selective gene mutations | fa/-, OSD * | Fat (timed +/−) | ✓ | Select gene mutants (fa/-) | N/A | ✓ | ✓ | Decr TC */HDL | ✓ |
| Diet (CHO:Fat:Protein %energy) | Diet 133 (60:20:20) * | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| T2DM ‘genetic permissiveness’ ranked by quintiles | Resist (10) | Resist (10) | Suscept (9) | Suscept (9) | Suscept (9) |
| RBG (range) after 10 weeks (mg/dL) | (48-61) | (62-69) | (71-143) | (183-424) | (427-600) |
| ave Random Blood Glucose (mg/dL) after 10weeks | 54 ± 5 ab | 66 ± 3 cd | 96 ± 27 ef | 269 ± 77 aceg | 535 ± 73 bdfg |
| Body Weight (g) after 10 weeks | 97 ± 9 ab | 102 ± 7 c | 105 ± 4 ad | 108 ± 4 be | 92 ± 10 cde |
| Food Intake in 9th week (kcal/d) | 31.8 ± 2.5 a | 34.2 ± 2.4 b | 34.7 ± 1.3 b | 35.2 ± 1.1 d | 43.8 ± 10.2 abcd |
| Water Intake in 9th week (mL/week) | 29 ± 7 a | 29 ± 7 b | 35 ± 10 c | 43 ± 22 d | 257 ± 289 abcd |
| Oral Glucose Tolerance Test (mg/dL) after 10 weeks | |||||
| Fasting Blood Glucose (FBG) 0 min | 50 ± 11 a | 49 ± 8 b | 47 ± 16 c | 44 ± 6 d | 92 ± 57 abcd |
| 30 min | 143 ± 49 abcd | 223 ± 58 ae | 257 ± 105 bf | 270 ± 49 cg | 490 ± 89 defg |
| Organ weight (%BW) | |||||
| Liver | 3.83 ± 0.67 a | 3.46 ± 0.17 b | 3.53 ± 0.22 c | 3.99 ± 0.76 d | 5.94 ± 1.17 abcd |
| Kidney | 0.70 ± 0.04 a | 0.68 ± 0.04 b | 0.68 ± 0.04 c | 0.78 ± 0.06 d | 1.22 ± 0.27 abcd |
| Cecum | 1.10 ± 0.42 a | 1.16 ± 0.21 b | 1.09 ± 0.17 c | 1.06 ± 0.19 d | 2.06 ± 0.65 abcd |
| Adipose | |||||
| Epididymal | 3.15 ± 0.70 a | 3.27 ± 0.39 b | 3.07 ± 0.50 c | 2.92 ± 0.69 d | 1.92 ± 0.86 abcd |
| Perirenal | 1.46 ± 0.33 abcd | 2.08 ± 0.52 ae | 2.03 ± 0.49 bf | 2.09 ± 0.48 cg | 0.68 ± 0.56 defg |
| Brown fat | 1.90 ± 0.39 abcd | 2.35 ± 0.57 ae | 2.68 ± 0.46 bf | 2.38 ± 0.43 cg | 1.10 ± 0.42 defg |
| Total fat | 6.51 ± 1.00 a | 7.01 ± 1.37 b | 7.42 ± 1.68 c | 6.66 ± 1.63 d | 3.70 ± 1.71 abcd |
| Plasma | |||||
| Cholesterol (mg/dL) | 126 ± 25 a | 124 ± 21 b | 125 ± 24 c | 145 ± 46 d | 482 ± 524 abcd |
| Triglycerides (mg/dL) | 63 ± 24 a | 71 ± 25 b | 73 ± 24 c | 110 ± 38 d | 292 ± 401 abcd |
| CHO:Fat:Protein %Energy | 70:10:20 | 70:10:20 | 70:10:20 | 70:10:20 | 70:10:20 | |||||
| Supplement | None | 0.075% StarlixTM (0.19 mg/kcal) | 0.05% AcarboseTM (0.13 mg/kcal) | 0.25% MetforminTM (0.63 mg/kcal) | 10% PFJ (1.4 mgGAE/kcal) ** | |||||
| 7 week RBG 75 mg/dL T2DM | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 |
| resist | suscept | resist | suscept | resist | suscept | resist | suscept | resist | suscept | |
| (n, %Incidence T2DM) | 5 | 7 (58%) | 4 | 8 (67%) | 4 | 1 (20%) | 5 | 1 (17%) | 10 | 2 (17%) |
| Random Body Weight (g) | ||||||||||
| Initial (3 weeks of age) | 31 ± 6 | 37 ± 4 abcd | 28 ± 7 a | 34 ± 4 | 36 ± 9 | 25 ± 0 | 31 ± 6 b | 23 ± 0 | 31 ± 5 c | 27 ± 1 d |
| After 4 weeks | 74 ± 9 | 72 ± 9 | 71 ± 14 | 78 ± 9 | 65 ± 5 | 77 ± 0 | 66 ± 4 | 79 ± 0 | 69 ± 8 | 69 ± 8 |
| After 7 weeks | 79 ± 19 | 83 ± 8 | 80 ± 14 | 86 ± 8 | 75 ± 6 a | 80 ± 0 | 84 ± 7 | 98 ± 0 | 88 ± 7 a | 83 ± 18 |
| Body Weight gain/day | 0.98 ± 0.37 | 0.94 ± 0.22 | 1.06 ± 0.27 | 1.06 ± 0.17 | 0.80 ± 0.26 | 1.12 ± 0.00 | 0.94 ± 0.06 | 1.19 ± 0.00 | 1.02 ± 0.13 | 1.00 ± 0.33 |
| After 4 weeks | 70 ± 17 ab | 178 ± 138 acdef | 73 ± 21 c | 199 ± 120 bhij | 79 ± 12 dh | 111 ± 0 | 70 ± 19 ei | 57 ± 0 | 74 ± 11 fj | 90 ± 14 |
| After 7 weeks | 61 ± 10 ab | 239 ± 171 acdefg | 58 ± 6 ch | 247 ± 197 bh | 63 ± 6 d | 104 ± 0 | 64 ± 8 e | 204 ± 0 | 55 ± 8 fg | 152 ± 85 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, A.; Landstrom, M.; Luu, A.; Hayes, K.C. The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM). Nutrients 2018, 10, 235. https://doi.org/10.3390/nu10020235
Subramaniam A, Landstrom M, Luu A, Hayes KC. The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM). Nutrients. 2018; 10(2):235. https://doi.org/10.3390/nu10020235
Chicago/Turabian StyleSubramaniam, Avinaash, Michelle Landstrom, Alice Luu, and K. C. Hayes. 2018. "The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM)" Nutrients 10, no. 2: 235. https://doi.org/10.3390/nu10020235
APA StyleSubramaniam, A., Landstrom, M., Luu, A., & Hayes, K. C. (2018). The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM). Nutrients, 10(2), 235. https://doi.org/10.3390/nu10020235