In Vivo Assessment of Resistant Starch Degradation by the Caecal Microbiota of Mice Using RNA-Based Stable Isotope Probing—A Proof-of-Principle Study

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Information
2.2. Feeding Experiment with Isotope-Labeled Raw Potato Starch
2.3. RNA Extraction, Isopycnic Ultracentrifugation, Gradient Fractionation, Reverse Transcription
2.4. Quantification of 16S rRNA in Gradient Fractions
2.5. 16S rRNA Amplicon Library Construction and Sequencing
2.6. 16S Ribosomal RNA Phylogenetic Analysis and Statistics
2.7. Organic Acid Analysis and Isotope Labeling Detection
3. Results
3.1. General Microbiota Structure in the Used Animals Prior to Gradient Separation
3.2. Density Gradient Formation and Recovery of Stable Isotope-Labeled Bacterial 16S rRNA
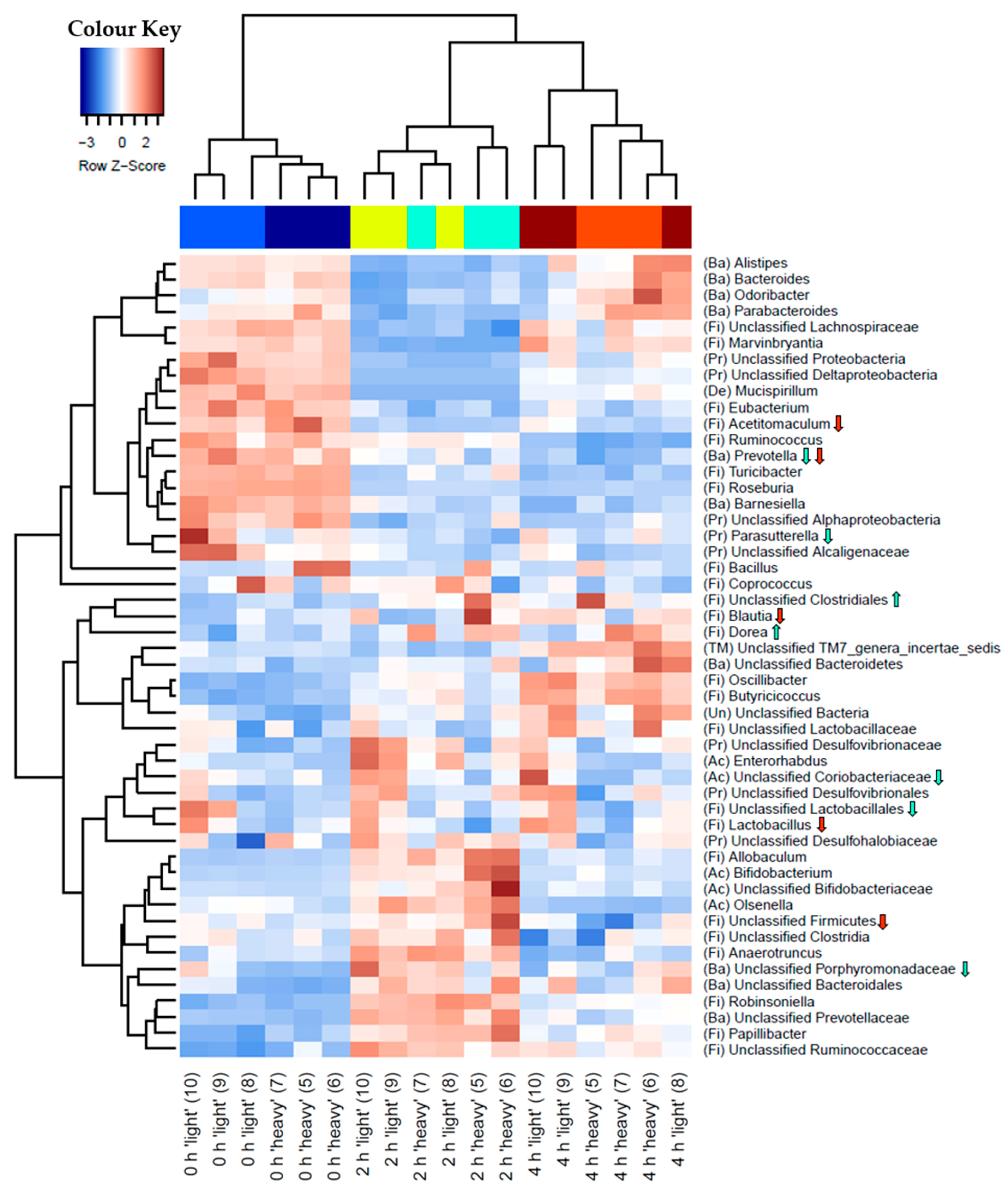
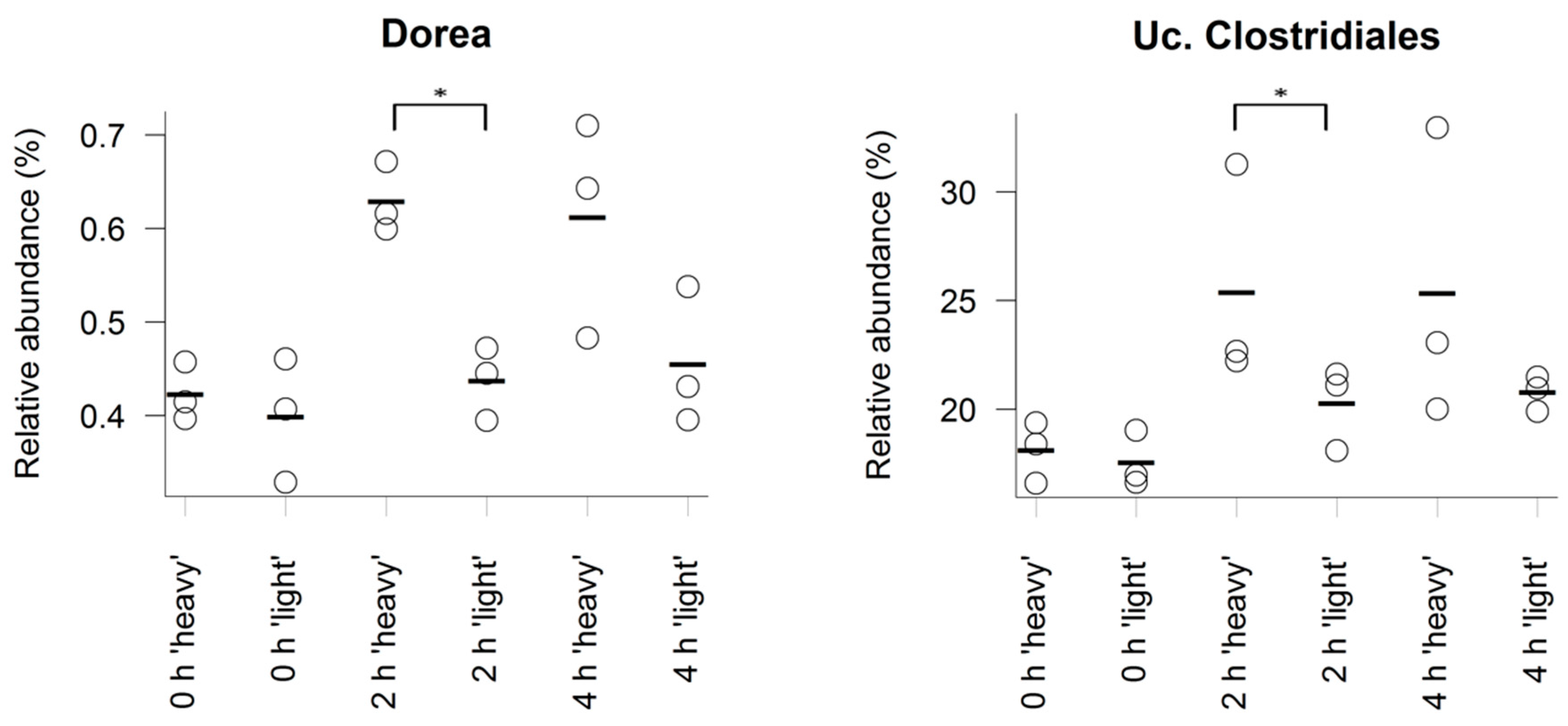
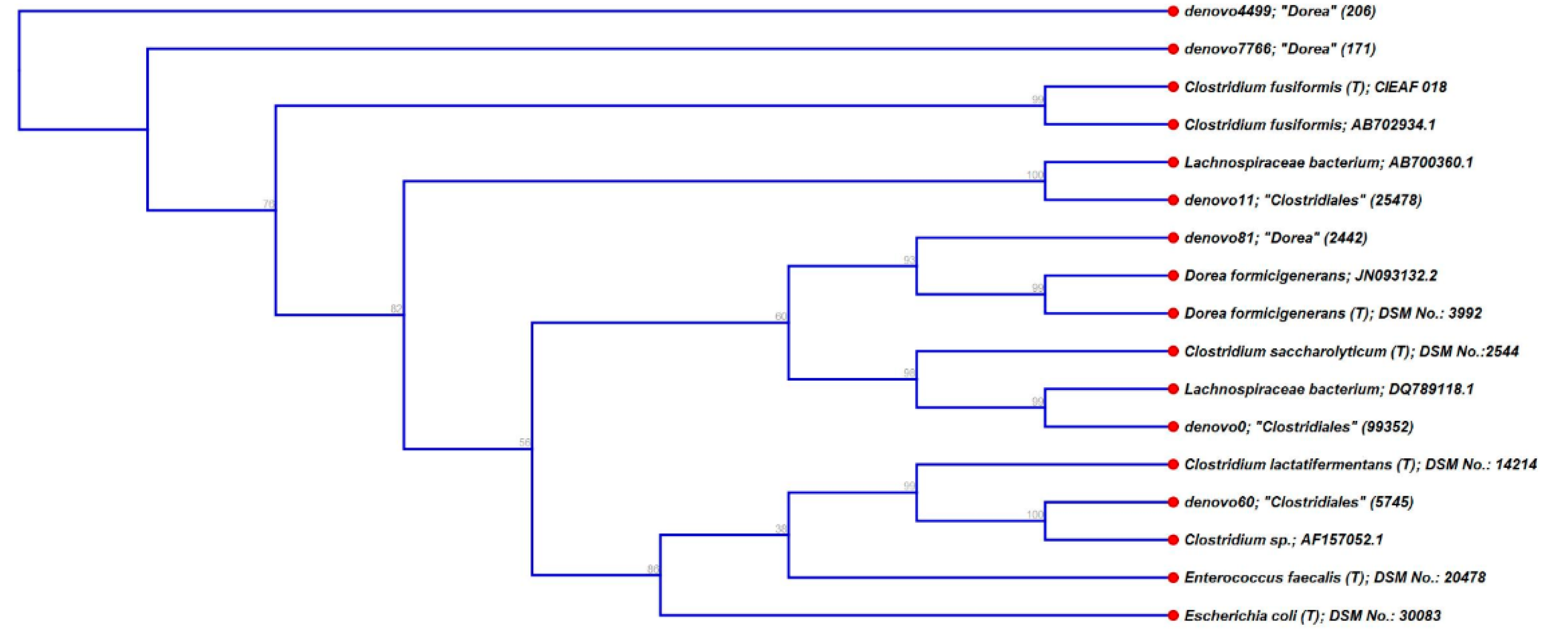
3.3. Characterisation of Metabolically Active Populations and Identification of Potential [U13C] Starch Degraders by Density Gradient Formation and Recovery of Stable Isotope-Labeled Bacterial 16S rRNA
3.4. Organic Acid Production and Heavy Isotope Incorporation into Metabolic Products
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Riedel, C.U.; Schwiertz, A.; Egert, M. The stomach and small and large intestinal microbiomes. In The Human Microbiota and Microbiome (Advances in Molecular and Cellular Microbiology 25); Marchesi, J.K., Ed.; CABI: Wallingford, UK, 2014; pp. 1–19. [Google Scholar]
- Turnbaugh, P.J.; Quince, C.; Faith, J.J.; McHardy, A.C.; Yatsunenko, T.; Niazi, F.; Affourtit, J.; Egholm, M.; Henrissat, B.; Knight, R.; et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. PNAS 2010, 107, 7503–7508. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- McNeil, N.I. The contribution of the large intestine to energy supplies in man. Am. J. Clin. Nutr. 1984, 39, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Asp, N.G. Resistant starch. Proceedings for the 2nd plenary meeting of Euresta: European flair concerted action No. 11 on physiological implications of the consumption of resistant starch in man. Crete, 29 May–2 June 1991. Eur. J. Clin. Nutr. 1992, 46 (Suppl. 2), S1–S48. [Google Scholar]
- Englyst, H.N.; Kingman, S.M.; Hudson, G.J.; Cummings, J.H. Measurement of resistant starch in vitro and in vivo. Br. J. Nutr. 1996, 75, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Birt, D.F.; Boylston, T.; Hendrich, S.; Jane, J.L.; Hollis, J.; Li, L.; McClelland, J.; Moore, S.; Phillips, G.J.; Rowling, M.; et al. Resistant starch: Promise for improving human health. Adv. Nutr. 2013, 4, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Englyst, H.N.; Kingman, S.M.; Cummings, J.H. Classification and measurement of nutritionally important starch fractions. Eur. J. Clin. Nutr. 1992, 46 (Suppl. 2), S33–S50. [Google Scholar] [PubMed]
- Slavin, J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients 2013, 5, 1417–1435. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.J.; Guinane, C.M.; O’Toole, P.W.; Cotter, P.D. Beneficial modulation of the gut microbiota. FEBS Lett. 2014, 588, 4120–4130. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.P. Health properties of resistant starch. BNF Nutr. Bull. 2005, 30, 27–54. [Google Scholar] [CrossRef]
- Higgins, J.A.; Brown, I.L. Resistant starch: A promising dietary agent for the prevention/treatment of inflammatory bowel disease and bowel cancer. Curr. Opin. Gastroenterol. 2013, 29, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Young, W.; Roy, N.C.; Lee, J.; Lawley, B.; Otter, D.; Henderson, G.; McCann, M.J.; Tannock, G.W. Changes in bowel microbiota induced by feeding weanlings resistant starch stimulate transcriptomic and physiological responses. Appl. Environ. Microbiol. 2012, 78, 6656–6664. [Google Scholar] [CrossRef] [PubMed]
- Kleessen, B.; Stoof, G.; Proll, J.; Schmiedl, D.; Noack, J.; Blaut, M. Feeding resistant starch affects fecal and cecal microflora and short-chain fatty acids in rats. J. Anim. Sci. 1997, 75, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Silvi, S.; Rumney, C.J.; Cresci, A.; Rowland, I.R. Resistant starch modifies gut microflora and microbial metabolism in human flora-associated rats inoculated with faeces from Italian and UK donors. J. Appl. Microbiol. 1999, 86, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Umu, O.C.; Frank, J.A.; Fangel, J.U.; Oostindjer, M.; da Silva, C.S.; Bolhuis, E.J.; Bosch, G.; Willats, W.G.; Pope, P.B.; Diep, D.B. Resistant starch diet induces change in the swine microbiome and a predominance of beneficial bacterial populations. Microbiome 2015, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Radajewski, S.; Ineson, P.; Parekh, N.R.; Murrell, J.C. Stable-isotope probing as a tool in microbial ecology. Nature 2000, 403, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Boschker, H.T.S.; Nold, S.C.; Wellsbury, P.; Bos, D.; de Graaf, W.; Pel, R.; Parkes, R.J.; Cappenberg, T.E. Direct linking of microbial populations to specific biogeochemical processes by 13C-labelling of biomarkers. Nature 1998, 392, 801–805. [Google Scholar] [CrossRef]
- Dumont, M.G.; Murrell, J.C. Stable isotope probing—Linking microbial identity to function. Nat. Rev. Microbiol. 2005, 3, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, J.D.; Dumont, M.G.; Vohra, J.; Murrell, J.C. Methodological considerations for the use of stable isotope probing in microbial ecology. Microb. Ecol. 2007, 53, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Egert, M.; de Graaf, A.A.; Smidt, H.; de Vos, W.M.; Venema, K. Beyond diversity: Functional microbiomics of the human colon. Trends Microbiol. 2006, 14, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Manefield, M.; Whiteley, A.S.; Ostle, N.; Ineson, P.; Bailey, M.J. Technical considerations for RNA-based stable isotope probing: An approach to associating microbial diversity with microbial community function. Rapid Commun. Mass Spectrom. 2002, 16, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Manefield, M.; Whiteley, A.S.; Griffiths, R.I.; Bailey, M.J. RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl. Environ. Microbiol. 2002, 68, 5367–5373. [Google Scholar] [CrossRef] [PubMed]
- Egert, M.; de Graaf, A.A.; Maathuis, A.; de Waard, P.; Plugge, C.M.; Smidt, H.; Deutz, N.E.; Dijkema, C.; de Vos, W.M.; Venema, K. Identification of glucose-fermenting bacteria present in an in vitro model of the human intestine by RNA-stable isotope probing. FEMS Microbial. Ecol. 2007, 60, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Kovatcheva-Datchary, P.; Egert, M.; Maathuis, A.; Rajilic-Stojanovic, M.; de Graaf, A.A.; Smidt, H.; de Vos, W.M.; Venema, K. Linking phylogenetic identities of bacteria to starch fermentation in an in vitro model of the large intestine by RNA-based stable isotope probing. Environ. Microbiol. 2009, 11, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W.; Lawley, B.; Munro, K.; Sims, I.M.; Lee, J.; Butts, C.A.; Roy, N. RNA-stable-isotope probing shows utilization of carbon from inulin by specific bacterial populations in the rat large bowel. Appl. Environ. Microbiol. 2014, 80, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Young, W.; Egert, M.; Bassett, S.A.; Bibiloni, R. Detection of sialic acid-utilising bacteria in a caecal community batch culture using RNA-based stable isotope probing. Nutrients 2015, 7, 2109–2124. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, E.; Young, W.; Rosendale, D.; Reichert-Grimm, V.; Riedel, C.U.; Conrad, R.; Egert, M. RNA-based stable isotope probing suggests Allobaculum spp. as particularly active glucose assimilators in a complex murine microbiota cultured in vitro. Biomed. Res. Int. 2017, 2017, 1829685. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, E.; Young, W.; Rosendale, D.; Conrad, R.; Riedel, C.U.; Egert, M. Determination of resistant starch assimilating bacteria in fecal samples of mice by in vitro RNA-based stable isotope probing. Front. Microbiol. 2017, 8, 1331. [Google Scholar] [CrossRef] [PubMed]
- Furet, J.P.; Firmesse, O.; Gourmelon, M.; Bridonneau, C.; Tap, J.; Mondot, S.; Dore, J.; Corthier, G. Comparative assessment of human and farm animal faecal microbiota using real-time quantitative PCR. FEMS Microbiol. Ecol. 2009, 68, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, E.; Koch, P.; Riedel, C.U.; Young, W.; Egert, M. Effect of rotor type on the separation of isotope-labeled and unlabeled Escherichia coli RNA by isopycnic density ultracentrifugation. Can. J. Microbiol. 2017, 63, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Lueders, T.; Manefield, M.; Friedrich, M.W. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ. Microbiol. 2004, 6, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; ISBN 3-900051-07-0. Available online: http://www.R-project.Org/ (accessed on 21 November 2017).
- Hervé, M. RVAideMemoire: Diverse Basic Statistical and Graphical Functions. R package version 0.9-45-2. 2015. Available online: https://cran.r-project.org/web/packages/RVAideMemoire/index.html (accessed on 1 February 2018).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Richardson, A.J.; Calder, A.G.; Stewart, C.S. Simultaneous determination of volatile and non-volatile acidic fermentation products of anaerobes by capillary Gas-Chromatography. Lett. Appl. Microbiol. 1989, 9, 5–8. [Google Scholar] [CrossRef]
- Slater, C.; Preston, T.; Weaver, L.T. Stable isotopes and the international system of units. Rapid Commun. Mass Spectrom. 2001, 15, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Tachon, S.; Zhou, J.; Keenan, M.; Martin, R.; Marco, M.L. The intestinal microbiota in aged mice is modulated by dietary resistant starch and correlated with improvements in host responses. FEMS Microbiol. Ecol. 2013, 83, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Tasman-Jones, C.; Englyst, H.; Harris, P.J. Comparative effects of three resistant starch preparations on transit time and short-chain fatty acid production in rats. Nutr. Cancer 2000, 36, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Charrier, J.A.; Martin, R.J.; McCutcheon, K.L.; Raggio, A.M.; Goldsmith, F.; Goita, M.; Senevirathne, R.N.; Brown, I.L.; Pelkman, C.; Zhou, J.; et al. High fat diet partially attenuates fermentation responses in rats fed resistant starch from high-amylose maize. Obesity (Silver Spring) 2013, 21, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, B.M.; Hildebrand, F.; Kubica, M.; Goosens, D.; Del Favero, J.; Declercq, W.; Raes, J.; Vandenabeele, P. Caspase deficiency alters the murine gut microbiome. Cell Death Dis. 2011, 2, e220. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, D.; Zhang, J.N.; Lv, X.; Wang, K.; Duan, L.P.; Nie, Y.; Wu, X.L. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 2013, 8, e74957. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, F.; Nguyen, T.L.; Brinkman, B.; Yunta, R.G.; Cauwe, B.; Vandenabeele, P.; Liston, A.; Raes, J. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013, 14, R4. [Google Scholar] [CrossRef] [PubMed]
- Salyers, A.A.; Vercellotti, J.R.; West, S.E.; Wilkins, T.D. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl. Environ. Microbiol. 1977, 33, 319–322. [Google Scholar] [PubMed]
- Salyers, A.A.; West, S.E.; Vercellotti, J.R.; Wilkins, T.D. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl. Environ. Microbiol. 1977, 34, 529–533. [Google Scholar] [PubMed]
- Macfarlane, G.T.; Englyst, H.N. Starch utilization by the human large intestinal microflora. J. Appl. Bacteriol. 1986, 60, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.C.; Koropatkin, N.M.; Smith, T.J.; Gordon, J.I. Complex glycan catabolism by the human gut microbiota: The Bacteroidetes sus-like paradigm. J. Biol. Chem. 2009, 284, 24673–24677. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Coyne, M.J.; Comstock, L.E. An ecological network of polysaccharide utilization among human intestinal symbionts. Curr. Biol. 2014, 24, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.L.; Avery, S.; Sen, P.; Swallow, D.M.; Hahn, D.; Sterchi, E. The maltase-glucoamylase gene: Common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc. Natl. Acad. Sci. USA 2003, 100, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Hino, S.; Ito, A.; Han, K.H.; Shimada, K.; Fukushima, M. Slower fermentation rate of potato starch relative to high-amylose cornstarch contributes to the higher proportion of cecal butyrate in rats. BMFH 2013, 32, 149–156. [Google Scholar] [CrossRef] [PubMed]
- De Rocha, T.S.; de Carneiro, A.P.A.; Franco, C.M.L. Effect of enzymatic hydrolysis on some physicochemical properties of root and tuber granular starches. Food Sci. Technol. (Campinas) 2010, 30, 544–551. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Jiminez, J.A.; Uwiera, T.C.; Abbott, D.W.; Uwiera, R.R.E.; Inglis, G.D. Impacts of resistant starch and wheat bran consumption on enteric inflammation in relation to colonic bacterial community structures and short-chain fatty acid concentrations in mice. Gut Pathog. 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Kim, J.; Duffy, P.R.; Schlegel, V.L.; Walter, J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS ONE 2010, 5, e15046. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, B.; McCormack, L.; Fardin-Kia, A.R.; Juenemann, R.; Nichenametla, S.; Clapper, J.; Specker, B.; Dey, M. Impact of dietary resistant starch type 4 on human gut microbiota and immunometabolic functions. Sci. Rep. 2016, 6, 28797. [Google Scholar] [CrossRef] [PubMed]
- Taras, D.; Simmering, R.; Collins, M.D.; Lawson, P.A.; Blaut, M. Reclassification of Eubacterium formicigenerans Holdeman and Moore 1974 as Dorea formicigenerans gen. nov., comb. nov., and description of Dorea longicatena sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2002, 52, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Bosshard, P.P.; Zbinden, R.; Altwegg, M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, gram-positive bacterium. Int. J. Syst. Evol. Microbiol. 2002, 52, 1263–1266. [Google Scholar] [PubMed]
- Van der Wielen, P.W.; Rovers, G.M.; Scheepens, J.M.; Biesterveld, S. Clostridium lactatifermentans sp. nov., a lactate-fermenting anaerobe isolated from the caeca of a chicken. Int. J. Syst. Evol. Microbiol. 2002, 52, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Su, Y.; Zhu, W. Microbiome-metabolome responses in the cecum and colon of pig to a high resistant starch diet. Front. Microbiol. 2016, 7, 779. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Giuberti, G.; Gallo, A.; Moschini, M.; Masoero, F. In vitro production of short-chain fatty acids from resistant starch by pig faecal inoculum. Animal 2013, 7, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Le Leu, R.K.; Hu, Y.; Brown, I.L.; Young, G.P. Effect of high amylose maize starches on colonic fermentation and apoptotic response to DNA-damage in the colon of rats. Nutr. Metab. (Lond.) 2009, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Le Leu, R.K.; Brown, I.L.; Hu, Y.; Morita, T.; Esterman, A.; Young, G.P. Effect of dietary resistant starch and protein on colonic fermentation and intestinal tumourigenesis in rats. Carcinogenesis 2007, 28, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Beatty, E.R.; Kingman, S.M.; Bingham, S.A.; Englyst, H.N. Digestion and physiological properties of resistant starch in the human large bowel. Br. J. Nutr. 1996, 75, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Dumon, H.J.; Lecannu, G.; Champ, M.M. Potato and high-amylose maize starches are not equivalent producers of butyrate for the colonic mucosa. Br. J. Nutr. 2000, 84, 689–696. [Google Scholar] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Flint, H.J. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 2004, 70, 5810–5817. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, A.; Duncan, S.H.; Calder, A.G.; Holtrop, G.; Louis, P.; Lobley, G.E.; Flint, H.J. Two routes of metabolic cross-feeding between bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl. Environ. Microbiol. 2006, 72, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Rios-Covian, D.; Gueimonde, M.; Duncan, S.H.; Flint, H.J.; de los Reyes-Gavilan, C.G. Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. FEMS Microbiol. Lett. 2015, 362, fnv176. [Google Scholar] [CrossRef] [PubMed]
- Hungate, B.A.; Mau, R.L.; Schwartz, E.; Caporaso, J.G.; Dijkstra, P.; van Gestel, N.; Koch, B.J.; Liu, C.M.; McHugh, T.A.; Marks, J.C.; et al. Quantitative microbial ecology through stable isotope probing. Appl. Environ. Microbiol. 2015, 81, 7570–7581. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Faith’s Phylogenetic Diversity Estimate | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| PD Whole Tree | |||||||||
| 0 h | 2 h | 4 h | p-Value | ||||||
| SIP Fractions | Mean | SEM | Mean | SEM | Mean | SEM | Density | Time | Density × Time |
| Heavy | 43.53 | 0.42 | 45.39 | 1.34 | 46.35 | 2.04 | 0.038 * | 0.023 * | 0.653 |
| Light | 46.86 | 0.38 | 45.02 | 0.47 | 50.92 | 1.01 | |||
| Fermentation Products | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration (μmol/mL Supernatant) | |||||||||
| 0 h | 2 h | 4 h | |||||||
| Organic acid (as conjugate base) | M1 | M2 | M3 | M4 | M5 | M6 | M7 | M8 | M9 |
| Formate (C1) | 0.3 a | 0.3 a | 0.3 a | 0.3 a | 0.3 a | 0.3 a | 0.3 a | 0.3 a | 0.3 a |
| Acetate (C2) | 17.77 | 16.6 | 18.7 | 18.7 | 13.39 | 6.69 | 10.82 | 7.27 | 14.22 |
| Propionate (C3) | 4.9 | 2.58 | 3.08 | 2.69 | 1.71 | 1.35 | 1.6 | 1.33 | 1.75 |
| Butyrate (C4) | 5.65 | 4.72 | 4.19 | 4.25 | 3.04 | 1.96 | 3.74 | 2.14 | 4.12 |
| Valerate (C5) | 0.23 | 0.17 | 0.17 | 0.26 | 0.22 | 0.13 | 0.16 | 0.12 | 0.18 |
| Caproate (C6) | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a |
| Enanthate (C7) | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a |
| Lactate (C3; 2-OH) | 0.59 | 0.96 | 3.66 | 0.94 | 0.38 | 0.8 | 0.67 | 0.25 | 0.48 |
| Succinate (C4; 1,4-dicarboxylate) | 0.52 | 0.45 | 0.82 | 0.47 | 0.32 | 0.31 | 0.33 | 0.3 a | 0.3 a |
| Isobutyrate (C4; 2-methyl-C3) | 0.25 | 0.15 a | 0.19 | 0.22 | 0.15 a | 0.15 a | 0.15 a | 0.15 a | 0.15 a |
| Isovalerate (C5; 3-methyl-C4) | 0.13 | 0.1 a | 0.14 | 0.13 | 0.1 a | 0.1 a | 0.1 a | 0.1 a | 0.1 a |
| 13C-Labeled Fermentation Products | ||||||
|---|---|---|---|---|---|---|
| 13C Atom Percent Excess (APE) Relative to Natural Abundance of 13C-Labeled Acid at Time 0 H Samples | ||||||
| 2 h | 4 h | |||||
| Organic acid (as conjugate base) | M4 | M5 | M6 | M7 | M8 | M9 |
| Acetate | 2.7 | 0.1 | 24.6 | 6.5 | 20.1 | 3.3 |
| Propionate | ND | ND | ND | ND | ND | ND |
| Butyrate | 6.4 | 0.4 | 39.7 | 14.8 | 38.3 | 8.0 |
| Lactate | 6.1 | 0.0 | 38.8 | 3.8 | 0.7 | 0.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, E.; Young, W.; Reichert-Grimm, V.; Weis, S.; Riedel, C.U.; Rosendale, D.; Stoklosinski, H.; Hunt, M.; Egert, M. In Vivo Assessment of Resistant Starch Degradation by the Caecal Microbiota of Mice Using RNA-Based Stable Isotope Probing—A Proof-of-Principle Study. Nutrients 2018, 10, 179. https://doi.org/10.3390/nu10020179
Herrmann E, Young W, Reichert-Grimm V, Weis S, Riedel CU, Rosendale D, Stoklosinski H, Hunt M, Egert M. In Vivo Assessment of Resistant Starch Degradation by the Caecal Microbiota of Mice Using RNA-Based Stable Isotope Probing—A Proof-of-Principle Study. Nutrients. 2018; 10(2):179. https://doi.org/10.3390/nu10020179
Chicago/Turabian StyleHerrmann, Elena, Wayne Young, Verena Reichert-Grimm, Severin Weis, Christian U. Riedel, Douglas Rosendale, Halina Stoklosinski, Martin Hunt, and Markus Egert. 2018. "In Vivo Assessment of Resistant Starch Degradation by the Caecal Microbiota of Mice Using RNA-Based Stable Isotope Probing—A Proof-of-Principle Study" Nutrients 10, no. 2: 179. https://doi.org/10.3390/nu10020179
APA StyleHerrmann, E., Young, W., Reichert-Grimm, V., Weis, S., Riedel, C. U., Rosendale, D., Stoklosinski, H., Hunt, M., & Egert, M. (2018). In Vivo Assessment of Resistant Starch Degradation by the Caecal Microbiota of Mice Using RNA-Based Stable Isotope Probing—A Proof-of-Principle Study. Nutrients, 10(2), 179. https://doi.org/10.3390/nu10020179