

Application of Macro X-ray Fluorescence Fast Mapping to Thickness Estimation of Layered Pigments




Abstract
1. Introduction
2. Research Aim
3. Materials and Methods
3.1. Stand-Alone Layers (Standard for Calibration)
3.2. Layered Painting Samples (Mockup)
- Sample A1: 37 ± 7.4 µm (azurite, upper layer) and 15 ± 7.4 µm (lead white)
- Sample A2: 22 ± 1.5 µm (ultramarine blue, upper layer) and 7.4 ± 7.4 µm (lead white)
- Sample A3: 7.5 ± 1.5 µm (ultramarine blue, upper layer), 18 ± 1.5 µm (azurite, intermediate layer) and 15 ± 7.4 µm (lead white).
3.3. MA-XRF Instrumentation
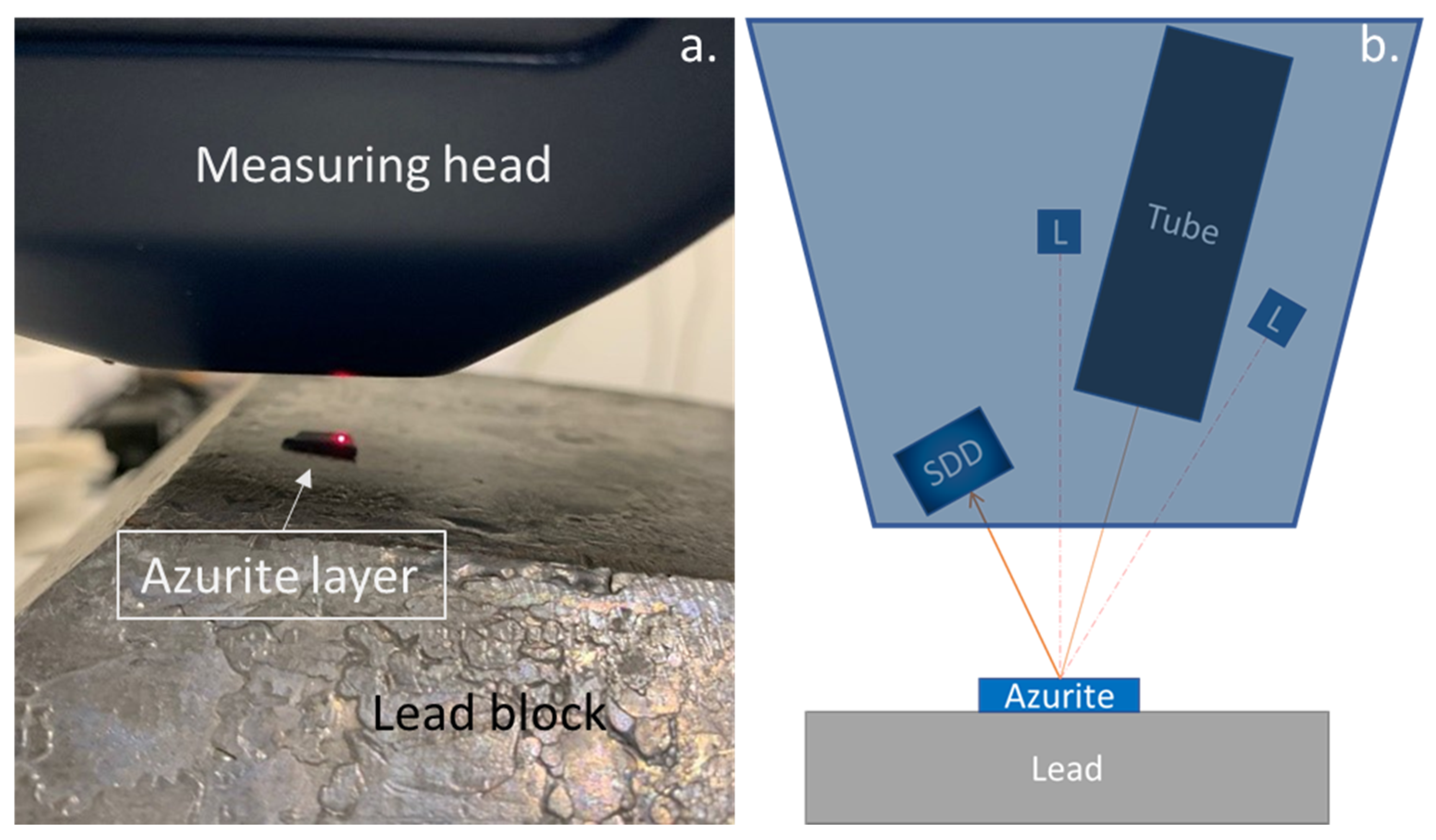
3.4. Feeler for Thickness Measurement of Stand-Alone Layers
4. Results and Discussion
- It is typically of no interest to evaluate the thickness over a specific pixel; instead, it is more interesting to look at the mean layer thickness, thus working with a cluster of pixels.
- Measuring time is too low to obtain reliable results over a specific pigment unless we consider more than one pixel, as real applications usually cannot perform multiple measurements on the same pixel to reach a good counting statistic.
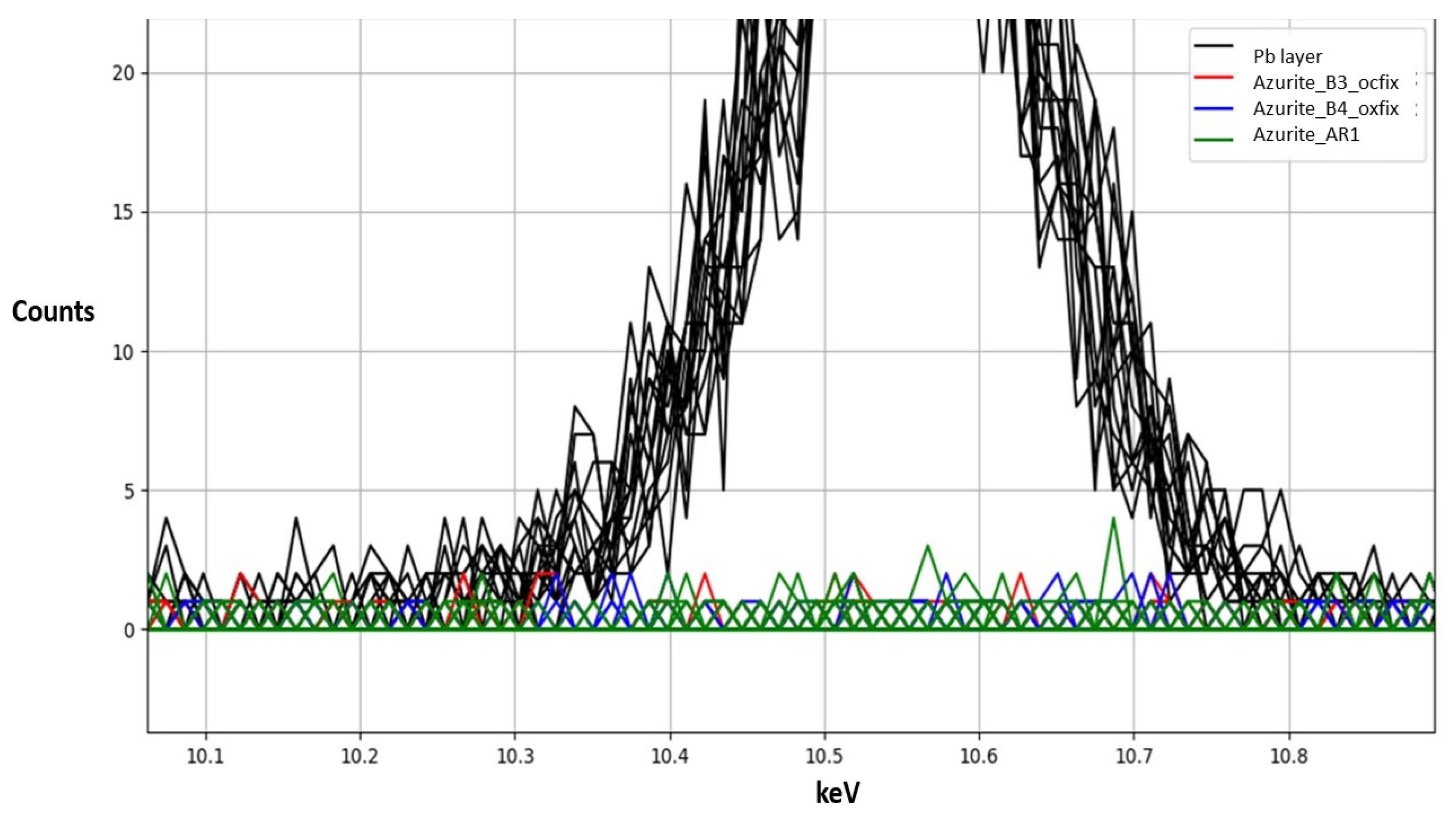
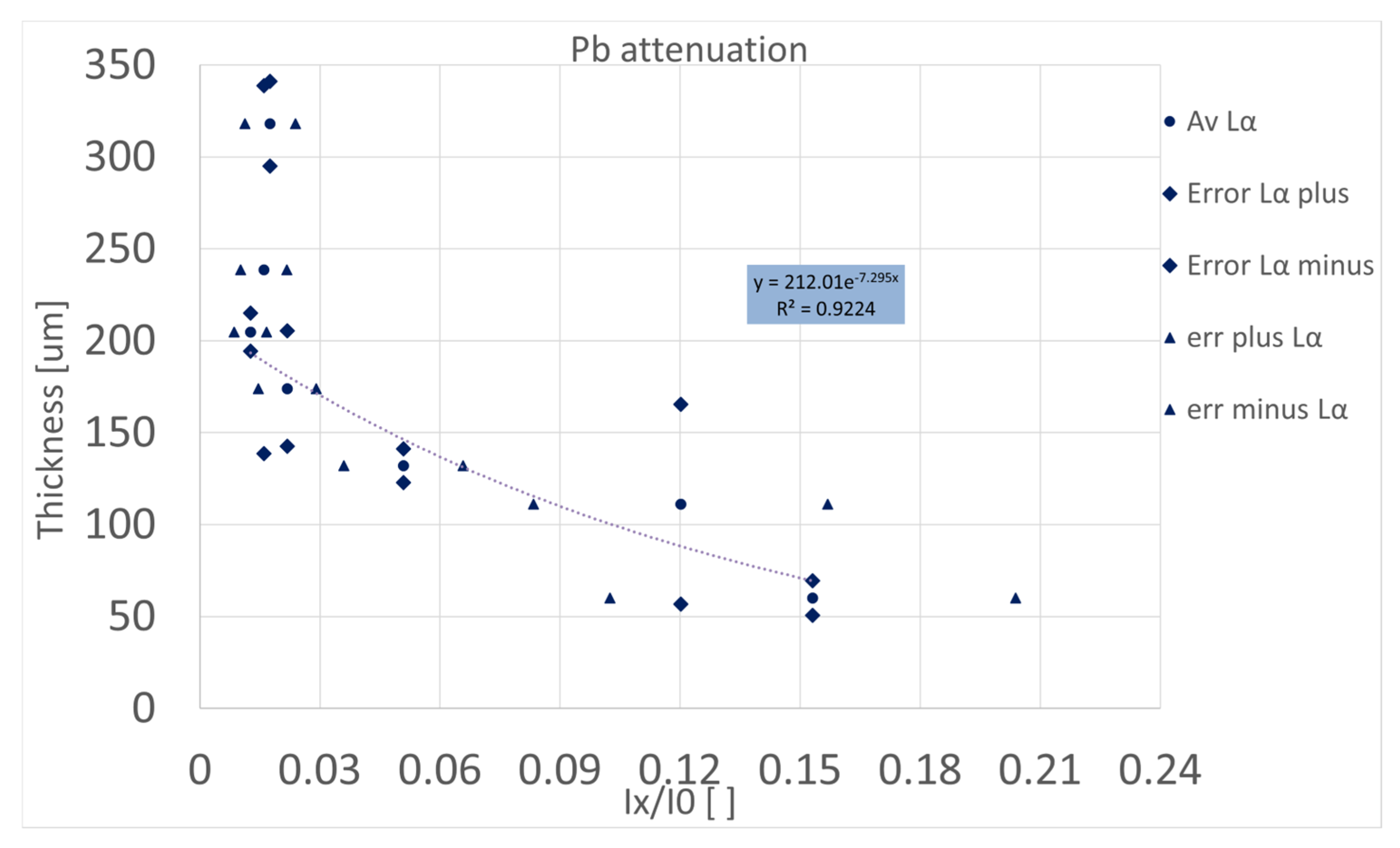
4.1. Thickness–Absorption Relation from Stand-Alone Layers
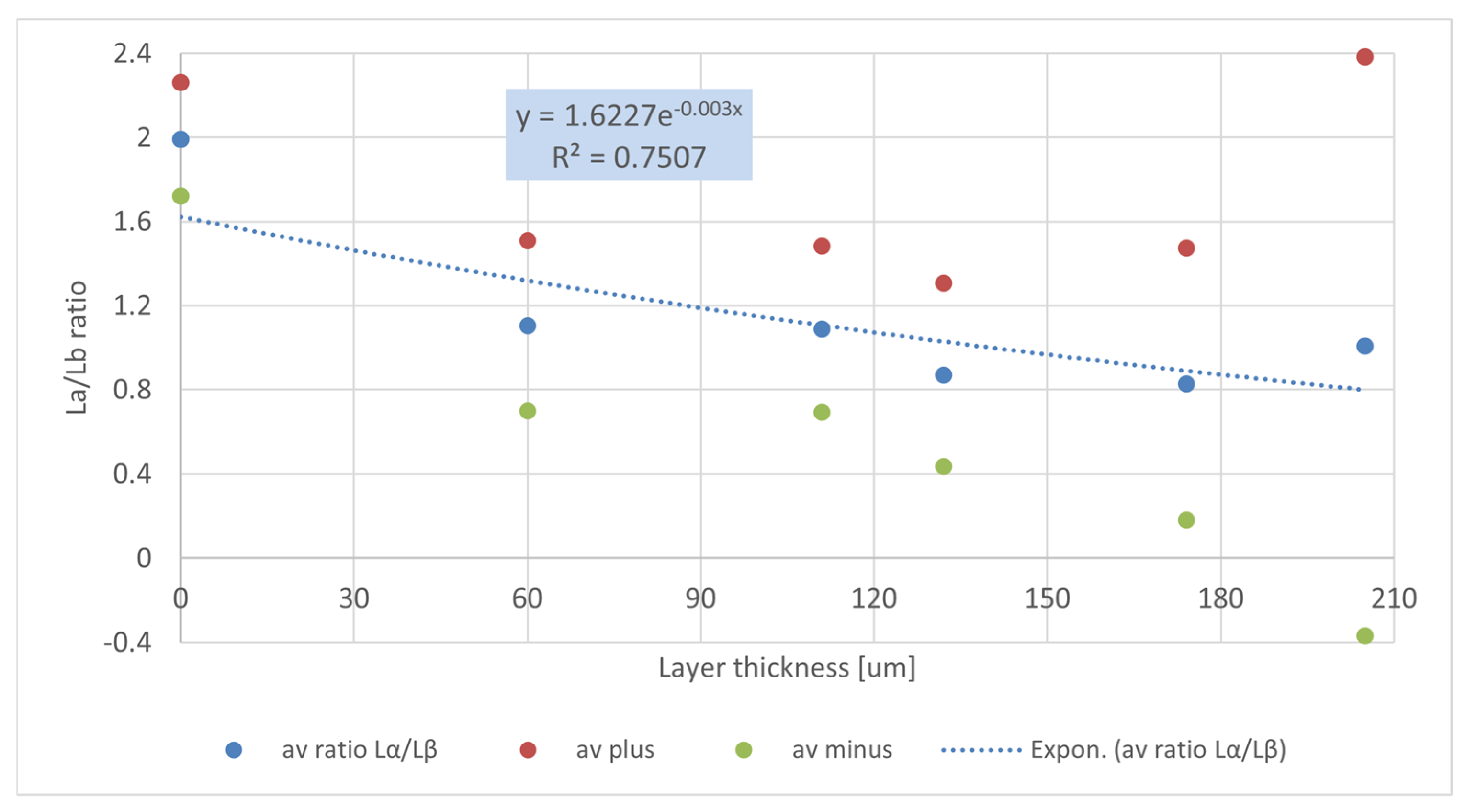
4.2. Calibration Curve
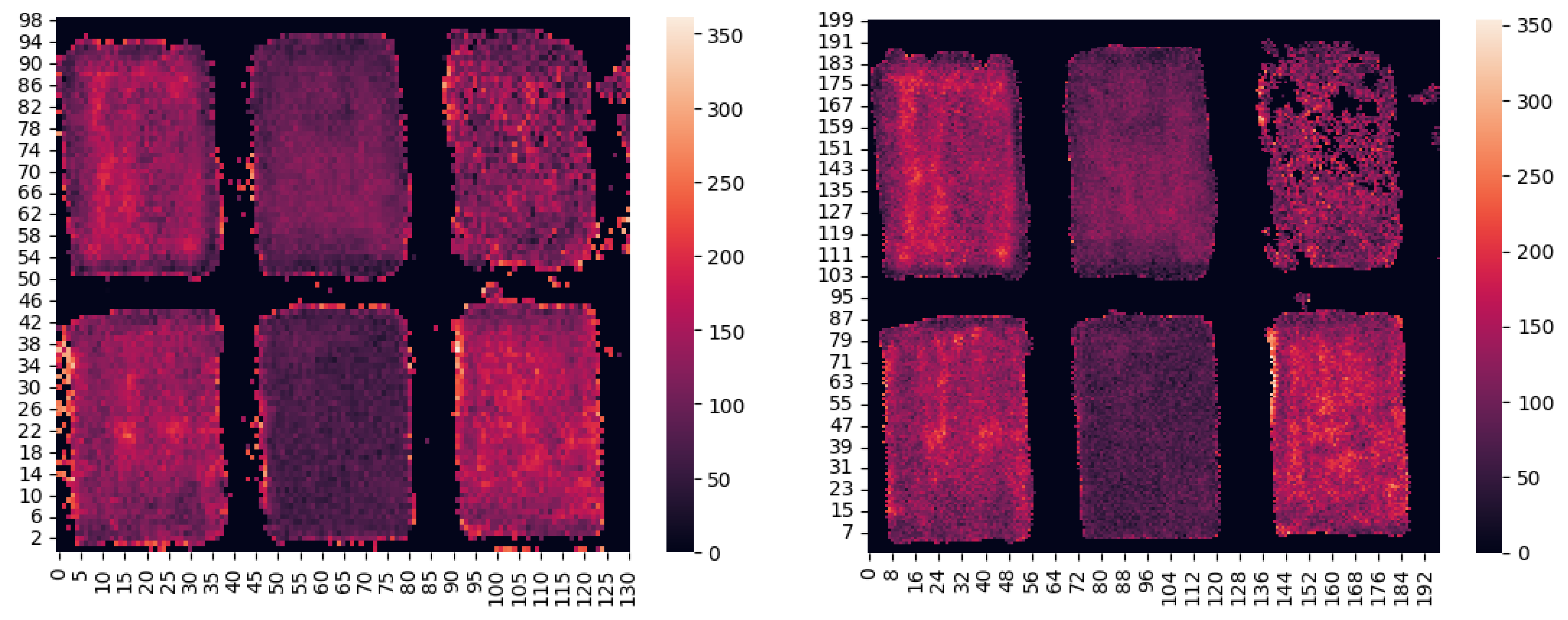
4.3. Thickness Maps of MOCKUP Layers
- The obtained image was transposed with respect to the actual object;
- The support is represented as a mid-thickness layer, which is incorrect.
- The information from the image was stored using the Python Pandas open-source library, exploiting the DataFrame built-in object. DataFrames are created as tables that collect keys in the form of [row][column]. On the contrary, the IRIS software creates a matrix of the type [column][row][spectrum], and populates it starting from the bottom left, scanning towards the right and ending at the top right. Therefore, it is necessary to lock the columns and range on the rows.
- Since the wood support presents noise in the lead line regions, due to backscattered radiation from the excitation source, the contrast can be highly increased considering a threshold to be overcome by at least one of the ROIs’ integrals. That threshold was set as the number of bins in the ROI times a constant of 1.1.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vadrucci, M.; Bazzano, G.; Borgognoni, F.; Chiari, M.; Mazzinghi, A.; Picardi, L.; Ronsivalle, C.; Ruberto, C.; Taccetti, F. A New Small-Footprint External-Beam PIXE Facility for Cultural Heritage Applications Using Pulsed Proton Beams. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2017, 406, 314–317. [Google Scholar] [CrossRef]
- Calligaro, T. PIXE in the Study of Archaeological and Historical Glass. X-ray Spectrom. 2008, 37, 169–177. [Google Scholar] [CrossRef]
- Sottili, L.; Giuntini, L.; Mazzinghi, A.; Massi, M.; Carraresi, L.; Castelli, L.; Czelusniak, C.; Giambi, F.; Mandò, P.A.; Manetti, M.; et al. The Role of PIXE and XRF in Heritage Science: The INFN-CHNet LABEC Experience. Appl. Sci. 2022, 12, 6585. [Google Scholar] [CrossRef]
- Moropoulou, A.; Zendri, E.; Ortiz, P.; Delegou, E.T.; Ntoutsi, I.; Balliana, E.; Becerra, J.; Ortiz, R. Scanning Microscopy Techniques as an Assessment Tool of Materials and Interventions for the Protection of Built Cultural Heritage. Scanning 2019, 2019, e5376214. [Google Scholar] [CrossRef]
- Calia, A.; Lettieri, M.; Quarta, G. Cultural Heritage Study: Microdestructive Techniques for Detection of Clay Minerals on the Surface of Historic Buildings. Appl. Clay Sci. 2011, 53, 525–531. [Google Scholar] [CrossRef]
- Ruvalcaba Sil, J.L.; Ramírez Miranda, D.; Aguilar Melo, V.; Picazo, F. SANDRA: A Portable XRF System for the Study of Mexican Cultural Heritage. X-ray Spectrom. 2010, 39, 338–345. [Google Scholar] [CrossRef]
- Liritzis, I.; Zacharias, N. Portable XRF of Archaeological Artifacts: Current Research, Potentials and Limitations. In X-ray Fluorescence Spectrometry (XRF) in Geoarchaeology; Shackley, M.S., Ed.; Springer: New York, NY, USA, 2011; pp. 109–142. ISBN 978-1-4419-6886-9. [Google Scholar]
- Andrić, V.; Gajić-Kvaščev, M.; Crkvenjakov, D.K.; Marić-Stojanović, M.; Gadžurić, S. Evaluation of Pattern Recognition Techniques for the Attribution of Cultural Heritage Objects Based on the Qualitative XRF Data. Microchem. J. 2021, 167, 106267. [Google Scholar] [CrossRef]
- Vaggelli, G.; Cossio, R. μ-XRF Analysis of Glasses: A Non-Destructive Utility for Cultural Heritage Applications. Analyst 2012, 137, 662–667. [Google Scholar] [CrossRef]
- Mantler, M.; Schreiner, M. X-ray Fluorescence Spectrometry in Art and Archaeology. X-ray Spectrom. 2000, 29, 3–17. [Google Scholar] [CrossRef]
- Dran, J.C.; Calligaro, T.; Salomon, J. Particle Induced X-ray Emission. In Modern Analytical Methods in Art and Arhaeology; Ciliberto, E., Spoto, G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2000; Volume 155, pp. 135–166. [Google Scholar]
- José-Yacaman, M.; Ascensio, J.A. Electron Microscopy and Ots Application to the Study of Archaeological Materials and Art Preservation. In Modern Analytical Methods in Art and Archaeology; Ciliberto, E., Spoto, G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2000; Volume 155, pp. 405–444. [Google Scholar]
- Bilo, F.; Cirelli, P.; Borgese, L. Elemental Analysis of Particulate Matter by X-ray Fluorescence Methods: A Green Approach to Air Quality Monitoring. TrAC Trends Anal. Chem. 2024, 170, 117427. [Google Scholar] [CrossRef]
- Chojnacka, K.; Mikulewicz, M. Green Analytical Methods of Metals Determination in Biosorption Studies. TrAC Trends Anal. Chem. 2019, 116, 254–265. [Google Scholar] [CrossRef]
- He, Y.; Tang, L.; Wu, X.; Hou, X.; Lee, Y. Spectroscopy: The Best Way Toward Green Analytical Chemistry? Appl. Spectrosc. Rev. 2007, 42, 119–138. [Google Scholar] [CrossRef]
- Balliana, E.; Ricci, G.; Pesce, C.; Zendri, E. Assessing the Value of Green Conservation for Cultural Heritage: Positive and Critical Aspects of Already Available Methodologies. Int. J. Conserv. Sci. 2016, 7, 185–202. [Google Scholar]
- Kučera, J.; Kameník, J.; Havránek, V.; Krausová, I.; Světlík, I.; Pachnerová Brabcová, K.; Fikrle, M.; Chvátil, D. Recent Achievements in NAA, PAA, XRF, IBA and AMS Applications for Cultural Heritage Investigations at Nuclear Physics Institute, Řež. Physics 2022, 4, 491–503. [Google Scholar] [CrossRef]
- NCSR “Demokritos” Institute of Nuclear and Particle Physics. Available online: http://www.inp.demokritos.gr/xrf/portable-xrf/ (accessed on 12 February 2024).
- Sri Lanka Atomic Energy Board. X-ray Fluorescence (XRF) Laboratory. Available online: https://aeb.gov.lk/x-ray-fluorescence-xrf-laboratory/ (accessed on 12 February 2024).
- International Atomic Energy Agency (IAEA). XRF-Training Program. Available online: https://nucleus.iaea.org/sites/nuclear-instrumentation/Pages/XRF-Training.aspx#:~:text=The%20training%20programs%20on%20XRF,from%208%20to%2014%20weeks.&text=Introduction%20to%20Quantitative%20analysis%3A%20Fundamentals,of%20possible%20approaches%20for%20quantification (accessed on 12 February 2024).
- IAEA. Final Report of a Coordinated Research Project 2000–2003: In Situ Applications of X-ray Fluorescence Techniques. Available online: https://www-pub.iaea.org/MTCD/Publications/PDF/te_1456_web.pdf (accessed on 12 February 2024).
- Macková, A.; MacGregor, D.; Azaiez, F.; Nyberg, J.; Piasetzky, E. Nuclear Physics for Cultural Heritage; Nuclear Physics Division of the European Physical Society: Mulhouse, France, 2016. [Google Scholar]
- Scruggs, B.; Haschke, M.; Herczeg, L.; Nicolosi, J. XRF Mapping: New Tools for Distribution Analysis. Adv. X-ray Anal. 2000, 42, 19–25. [Google Scholar]
- Campos, P.H.O.V.; Appoloni, C.R.; Rizzutto, M.A.; Leite, A.R.; Assis, R.F.; Santos, H.C.; Silva, T.F.; Rodrigues, C.L.; Tabacniks, M.H.; Added, N. A Low-Cost Portable System for Elemental Mapping by XRF Aiming In Situ Analyses. Appl. Radiat. Isot. 2019, 152, 78–85. [Google Scholar] [CrossRef]
- Haschke, M.; Rossek, U.; Tagle, R.; Waldschläger, U. Fast elemental mapping with micro-XRF. Adv X-ray Anal. 2012, 55, 286–298. [Google Scholar]
- Alfeld, M. MA-XRF for Historical Paintings: State of the Art and Perspective. Microsc. Microanal. 2020, 26, 72–75. [Google Scholar] [CrossRef]
- Galli, A.; Caccia, M.; Alberti, R.; Bonizzoni, L.; Aresi, N.; Frizzi, T.; Bombelli, L.; Gironda, M.; Martini, M. Discovering the Material Palette of the Artist: A p-XRF Stratigraphic Study of the Giotto Panel ‘God the Father with Angels’. X-ray Spectrom. 2017, 46, 435–441. [Google Scholar] [CrossRef]
- Gu, B.; Mishra, B.; Miller, C.; Wang, W.; Lai, B.; Brooks, S.C.; Kemner, K.M.; Liang, L. X-ray Fluorescence Mapping of Mercury on Suspended Mineral Particles and Diatoms in a Contaminated Freshwater System. Biogeosciences 2014, 11, 5259–5267. [Google Scholar] [CrossRef]
- Alberti, R.; Frizzi, T.; Bombelli, L.; Gironda, M.; Aresi, N.; Rosi, F.; Miliani, C.; Tranquilli, G.; Talarico, F.; Cartechini, L. CRONO: A Fast and Reconfigurable Macro X-ray Fluorescence Scanner for in-Situ Investigations of Polychrome Surfaces. X-ray Spectrom. 2017, 46, 297–302. [Google Scholar] [CrossRef]
- Pouyet, E.; Barbi, N.; Chopp, H.; Healy, O.; Katsaggelos, A.; Moak, S.; Mott, R.; Vermeulen, M.; Walton, M. Development of a Highly Mobile and Versatile Large MA-XRF Scanner for in Situ Analyses of Painted Work of Arts. X-ray Spectrom. 2021, 50, 263–271. [Google Scholar] [CrossRef]
- Mazzinghi, A.; Ruberto, C.; Castelli, L.; Ricciardi, P.; Czelusniak, C.; Giuntini, L.; Mandò, P.A.; Manetti, M.; Palla, L.; Taccetti, F. The Importance of Being Little: MA-XRF on Manuscripts on a Venetian Island. X-ray Spectrom. 2021, 50, 272–278. [Google Scholar] [CrossRef]
- Ruberto, C.; Mazzinghi, A.; Massi, M.; Castelli, L.; Czelusniak, C.; Palla, L.; Gelli, N.; Betuzzi, M.; Impallaria, A.; Brancaccio, R.; et al. Imaging Study of Raffaello’s “La Muta” by a Portable XRF Spectrometer. Microchem. J. 2016, 126, 63–69. [Google Scholar] [CrossRef]
- Taccetti, F.; Castelli, L.; Czelusniak, C.; Gelli, N.; Mazzinghi, A.; Palla, L.; Ruberto, C.; Censori, C.; Lo Giudice, A.; Re, A.; et al. A Multipurpose X-ray Fluorescence Scanner Developed for in Situ Analysis. Rend. Fis. Acc. Lincei 2019, 30, 307–322. [Google Scholar] [CrossRef]
- Lins, S.A.B.; Manso, M.; Lins, P.A.B.; Brunetti, A.; Sodo, A.; Gigante, G.E.; Fabbri, A.; Branchini, P.; Tortora, L.; Ridolfi, S. Modular MA-XRF Scanner Development in the Multi-Analytical Characterisation of a 17th Century Azulejo from Portugal. Sensors 2021, 21, 1913. [Google Scholar] [CrossRef] [PubMed]
- Orsilli, J.; Galli, A.; Bonizzoni, L.; Caccia, M. More than XRF Mapping: STEAM (Statistically Tailored Elemental Angle Mapper) a Pioneering Analysis Protocol for Pigment Studies. Appl. Sci. 2021, 11, 1446. [Google Scholar] [CrossRef]
- Vanhoof, C.; Bacon, J.R.; Fittschen, U.E.A.; Vincze, L. Atomic Spectrometry Update—A Review of Advances in X-ray Fluorescence Spectrometry and Its Special Applications. J. Anal. At. Spectrom. 2021, 36, 1797–1812. [Google Scholar] [CrossRef]
- Dos Santos, H.C.; Caliri, C.; Pappalardo, L.; Catalano, R.; Orlando, A.; Rizzo, F.; Romano, F.P. Real-Time MA-XRF Imaging Spectroscopy of the Virgin with the Child Painted by Antonello de Saliba in 1497. Microchem. J. 2018, 140, 96–104. [Google Scholar] [CrossRef]
- Romano, F.P.; Caliri, C.; Nicotra, P.; Martino, S.D.; Pappalardo, L.; Rizzo, F.; Santos, H.C. Real-Time Elemental Imaging of Large Dimension Paintings with a Novel Mobile Macro X-ray Fluorescence (MA-XRF) Scanning Technique. J. Anal. At. Spectrom. 2017, 32, 773–781. [Google Scholar] [CrossRef]
- Sciutto, G.; Frizzi, T.; Catelli, E.; Aresi, N.; Prati, S.; Alberti, R.; Mazzeo, R. From Macro to Micro: An Advanced Macro X-ray Fluorescence (MA-XRF) Imaging Approach for the Study of Painted Surfaces. Microchem. J. 2018, 137, 277–284. [Google Scholar] [CrossRef]
- Saverwyns, S.; Currie, C.; Lamas-Delgado, E. Macro X-ray Fluorescence Scanning (MA-XRF) as Tool in the Authentication of Paintings. Microchem. J. 2018, 137, 139–147. [Google Scholar] [CrossRef]
- Ricciardi, P.; Legrand, S.; Bertolotti, G.; Janssens, K. Macro X-ray Fluorescence (MA-XRF) Scanning of Illuminated Manuscript Fragments: Potentialities and Challenges. Microchem. J. 2016, 124, 785–791. [Google Scholar] [CrossRef]
- Cavaleri, T.; Pelosi, C.; Ricci, M.; Laureti, S.; Romano, F.P.; Caliri, C.; Ventura, B.; De Blasi, S.; Gargano, M. IR Reflectography, Pulse-Compression Thermography, MA-XRF, and Radiography: A Full-Thickness Study of a 16th-Century Panel Painting Copy of Raphael. J. Imaging 2022, 8, 150. [Google Scholar] [CrossRef]
- Bicchieri, M.; Biocca, P.; Caliri, C.; Romano, F.P. Complementary MA-XRF and μ-Raman Results on Two Leonardo Da Vinci Drawings. X-ray Spectrom. 2021, 50, 401–409. [Google Scholar] [CrossRef]
- Kogou, S.; Lee, L.; Shahtahmassebi, G.; Liang, H. A New Approach to the Interpretation of XRF Spectral Imaging Data Using Neural Networks. X-ray Spectrom. 2021, 50, 310–319. [Google Scholar] [CrossRef]
- Bonizzoni, L.; Galli, A.; Poldi, G.; Milazzo, M. In Situ Non-Invasive EDXRF Analysis to Reconstruct Stratigraphy and Thickness of Renaissance Pictorial Multilayers. X-ray Spectrom. 2007, 36, 55–61. [Google Scholar] [CrossRef]
- Trojek, T.; Wegrzynek, D. X-ray Fluorescence Kα/Kβ Ratios for a Layered Specimen: Comparison of Measurements and Monte Carlo Calculations with the MCNPX Code. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2010, 619, 311–315. [Google Scholar] [CrossRef]
- Fiorini, C.; Gianoncelli, A.; Longoni, A.; Zaraga, F. Determination of the Thickness of Coatings by Means of a New XRF Spectrometer. X-ray Spectrom. 2002, 31, 92–99. [Google Scholar] [CrossRef]
- Vavrik, D.; Kytyr, D.; Zemlicka, J. Stratigraphy of a Layered Structure Utilizing XRF and Scattered Photons. J. Inst. 2020, 15, C03011. [Google Scholar] [CrossRef]
- Bonizzoni, L.; Colombo, C.; Ferrati, S.; Gargano, M.; Greco, M.; Ludwig, N.; Realini, M. A Critical Analysis of the Application of EDXRF Spectrometry on Complex Stratigraphies. X-ray Spectrom. 2011, 40, 247–253. [Google Scholar] [CrossRef]
- Orsilli, J.; Migliori, A.; Padilla-Alvarez, R.; Martini, M.; Galli, A. AR-XRF Measurements and Data Treatment for the Evaluation of Gilding Samples of Cultural Heritage. J. Anal. At. Spectrom. 2023, 38, 174–185. [Google Scholar] [CrossRef]
- Orsilli, J.; Martini, M.; Galli, A. Angle Resolved-XRF Analysis of Puebla Ceramic Decorations. Spectrochim. Acta Part B At. Spectrosc. 2023, 210, 106809. [Google Scholar] [CrossRef]
- Liang, H.; Lucian, A.; Lange, R.; Cheung, C.S.; Su, B. Remote Spectral Imaging with Simultaneous Extraction of 3D Topography for Historical Wall Paintings. ISPRS J. Photogramm. Remote Sens. 2014, 95, 13–22. [Google Scholar] [CrossRef]
- Förste, F.; Bauer, L.; Heimler, K.; Hansel, B.; Vogt, C.; Kanngießer, B.; Mantouvalou, I. Quantification Routines for Full 3D Elemental Distributions of Homogeneous and Layered Samples Obtained with Laboratory Confocal Micro XRF Spectrometers. J. Anal. At. Spectrom. 2022, 37, 1687–1695. [Google Scholar] [CrossRef]
- Liang, J. Mixing Worlds: Current Trends in Integrating the Past and Present through Augmented and Mixed Reality. Adv. Archaeol. Pract. 2021, 9, 250–256. [Google Scholar] [CrossRef]
- Gettens, R.J.; Kühn, H.; Chase, W.T. Lead White. In Artists’ Pigments; Roy, A., Ed.; National Gallery of Art: Washington, DC, USA, 1993; Volume 2, pp. 67–82. [Google Scholar]
- Gargano, M.; Galli, A.; Bonizzoni, L.; Alberti, R.; Aresi, N.; Caccia, M.; Castiglioni, I.; Interlenghi, M.; Salvatore, C.; Ludwig, N.; et al. The Giotto’s Workshop in the XXI Century: Looking inside the “God the Father with Angels” Gable. J. Cult. Herit. 2019, 36, 255–263. [Google Scholar] [CrossRef]
- Special Engineering. Available online: https://www.bruker.com/en/applications/academia-materials-science/art-conservation-archaeology/special-engineering.html (accessed on 21 September 2023).
- Occhipinti, M.; Alberti, R.; Parsani, T.; Dicorato, C.; Tirelli, P.; Gironda, M.; Tocchio, A.; Frizzi, T. IRIS: A Novel Integrated Instrument for Co-Registered MA-XRF Mapping and VNIR-SWIR Hyperspectral Imaging. X-ray Spectrom. 2023. [Google Scholar] [CrossRef]
- Gargano, M.; Bonizzoni, L.; Grifoni, E.; Melada, J.; Guglielmi, V.; Bruni, S.; Ludwig, N. Multi-Analytical Investigation of Panel, Pigments and Varnish of The Martyirdom of St. Catherine by Gaudenzio Ferrari (16th Century). J. Cult. Herit. 2020, 46, 289–297. [Google Scholar] [CrossRef]
- Bonizzoni, L.; Gargano, M.; Ludwig, N.; Martini, M.; Galli, A. Looking for Common Fingerprints in Leonardo’s Pupils Using Nondestructive Pigment Characterization. Appl. Spectrosc. 2017, 71, 1915–1926. [Google Scholar] [CrossRef]
- Galli, A.; Poldi, G.; Martini, M.; Sibilia, E.; Montanari, C.; Panzeri, L. Study of Blue Colour in Ancient Mosaic Tesserae by Means of Thermoluminescence and Reflectance Measurements. Appl. Phys. A 2006, 83, 675–679. [Google Scholar] [CrossRef]
- Micheletti, F.; Orsilli, J.; Melada, J.; Gargano, M.; Ludwig, N.; Bonizzoni, L. The Role of IRT in the Archaeometric Study of Ancient Glass through XRF and FORS. Microchem. J. 2020, 153, 104388. [Google Scholar] [CrossRef]
- Gettens, R.J.; Kühn, H.; Chase, W.T. 3. Lead White. Stud. Conserv. 1967, 12, 125–139. [Google Scholar] [CrossRef]
- Morgan, S.; Townsend, J.H.; Hackney, S.; Perry, R. Canvas and Its Preparation in Early Twentieth-Century British Paintings. In The Camden Town Group in Context; Tate: London, UK, 2012; ISBN 978-1-84976-385-1. [Google Scholar]
- Gettens, R.J.; Mrose, M.E. Calcium Sulphate Minerals in the Grounds of Italian Paintings. Stud. Conserv. 1954, 1, 174–189. [Google Scholar] [CrossRef]
- De Viguerie, L.; Glanville, H.; Ducouret, G.; Jacquemot, P.; Dang, P.A.; Walter, P. Re-Interpretation of the Old Masters’ Practices through Optical and Rheological Investigation: The Presence of Calcite. Comptes Rendus Phys. 2018, 19, 543–552. [Google Scholar] [CrossRef]
- Dance, D.R.; Christofides, S.; Maidment, A.D.A.; McLean, J.D.; Ng, K.H. Chapter 2: Interactions of Radiation with Matter Diagnostic Radiology. In Physics: A Handbook for Teachers and Students; IAEA: Vienna, Austria, 2014; pp. 24–28. [Google Scholar]
- Cesareo, R.; Rizzutto, M.A.; Brunetti, A.; Rao, D.V. Metal Location and Thickness in a Multilayered Sheet by Measuring Kα/Kβ, Lα/Lβ and Lα/Lγ X-ray Ratios. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2009, 267, 2890–2896. [Google Scholar] [CrossRef]
- Cesareo, R.; De Assis, J.T.; Roldán, C.; Bustamante, A.D.; Brunetti, A.; Schiavon, N. Multilayered Samples Reconstructed by Measuring Kα/Kβ or Lα/Lβ X-ray Intensity Ratios by EDXRF. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2013, 312, 15–22. [Google Scholar] [CrossRef]
- Bonizzoni, L.; Galli, A.; Poldi, G. In Situ EDXRF Analyses on Renaissance Plaquettes and Indoor Bronzes Patina Problems and Provenance Clues. X-ray Spectrom. 2008, 37, 388–394. [Google Scholar] [CrossRef]
- Buccolieri, G.; Buccolieri, A.; Donati, P.; Marabelli, M.; Castellano, A. Portable EDXRF Investigation of the Patinas on the Riace Bronzes. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2015, 343, 101–109. [Google Scholar] [CrossRef]
- Garmay, A.V.; Oskolok, K.V.; Monogarova, O.V. μXRF Analysis of XVIII Century Copper Coin: Patina Investigation and “Bronze Disease” Detection. Mosc. Univ. Chem. Bull. 2021, 76, 133–136. [Google Scholar] [CrossRef]
- Schmidt, C.M.; Walton, M.S.; Trentelman, K. Characterization of Lapis Lazuli Pigments Using a Multitechnique Analytical Approach: Implications for Identification and Geological Provenancing. Anal. Chem. 2009, 81, 8513–8518. [Google Scholar] [CrossRef]
- Sitko, R. Quantitative X-ray Fluorescence Analysis of Samples of Less than ‘Infinite Thickness’: Difficulties and Possibilities. Spectrochim. Acta Part B At. Spectrosc. 2009, 64, 1161–1172. [Google Scholar] [CrossRef]
- Dupuis, G.; Elias, M.; Simonot, L. Pigment Identification by Fiber-Optics Diffuse Reflectance Spectroscopy. Appl. Spectrosc. 2002, 56, 1329–1336. [Google Scholar] [CrossRef]
- Miliani, C.; Rosi, F.; Daveri, A.; Brunetti, B.G. Reflection Infrared Spectroscopy for the Non-Invasive In Situ Study of Artists’ Pigments. Appl. Phys. A 2012, 106, 295–307. [Google Scholar] [CrossRef]
- Dupuis, G.; Menu, M. Quantitative Characterisation of Pigment Mixtures Used in Art by Fibre-Optics Diffuse-Reflectance Spectroscopy. Appl. Phys. A 2006, 83, 469–474. [Google Scholar] [CrossRef]
- De Viguerie, L.; Rochut, S.; Alfeld, M.; Walter, P.; Astier, S.; Gontero, V.; Boulc’h, F. XRF and Reflectance Hyperspectral Imaging on a 15th Century Illuminated Manuscript: Combining Imaging and Quantitative Analysis to Understand the Artist’s Technique. Herit. Sci. 2018, 6, 11. [Google Scholar] [CrossRef]
- Hradil, D.; Bezdička, P.; Hradilová, J.; Vašutová, V. Microanalysis of Clay-Based Pigments in Paintings by XRD Techniques. Microchem. J. 2016, 125, 10–20. [Google Scholar] [CrossRef]
- Franquelo, M.L.; Duran, A.; Castaing, J.; Arquillo, D.; Perez-Rodriguez, J.L. XRF, μ-XRD and μ-Spectroscopic Techniques for Revealing the Composition and Structure of Paint Layers on Polychrome Sculptures after Multiple Restorations. Talanta 2012, 89, 462–469. [Google Scholar] [CrossRef]
- Brostoff, L.B.; Centeno, S.A.; Ropret, P.; Bythrow, P.; Pottier, F. Combined X-ray Diffraction and Raman Identification of Synthetic Organic Pigments in Works of Art: From Powder Samples to Artists’ Paints. Anal. Chem. 2009, 81, 6096–6106. [Google Scholar] [CrossRef]
- Salvadó, N.; Butí, S.; Aranda, M.A.G.; Pradell, T. New Insights on Blue Pigments Used in 15th Century Paintings by Synchrotron Radiation-Based Micro-FTIR and XRD. Anal. Methods 2014, 6, 3610–3621. [Google Scholar] [CrossRef]
- Herrera, L.K.; Cotte, M.; Jimenez de Haro, M.C.; Duran, A.; Justo, A.; Perez-Rodriguez, J.L. Characterization of Iron Oxide-Based Pigments by Synchrotron-Based Micro X-ray Diffraction. Appl. Clay Sci. 2008, 42, 57–62. [Google Scholar] [CrossRef]
- Appolonia, L.; Vaudan, D.; Chatel, V.; Aceto, M.; Mirti, P. Combined Use of FORS, XRF and Raman Spectroscopy in the Study of Mural Paintings in the Aosta Valley (Italy). Anal. Bioanal. Chem. 2009, 395, 2005–2013. [Google Scholar] [CrossRef]
- Paternoster, G.; Rinzivillo, R.; Nunziata, F.; Castellucci, E.M.; Lofrumento, C.; Zoppi, A.; Felici, A.C.; Fronterotta, G.; Nicolais, C.; Piacentini, M.; et al. Study on the Technique of the Roman Age Mural Paintings by Micro-XRF with Polycapillary Conic Collimator and Micro-Raman Analyses. J. Cult. Herit. 2005, 6, 21–28. [Google Scholar] [CrossRef]
- Madariaga, J.M.; Maguregui, M.; De Vallejuelo, S.F.-O.; Knuutinen, U.; Castro, K.; Martinez-Arkarazo, I.; Giakoumaki, A.; Pitarch, A. In Situ Analysis with Portable Raman and ED-XRF Spectrometers for the Diagnosis of the Formation of Efflorescence on Walls and Wall Paintings of the Insula IX 3 (Pompeii, Italy). J. Raman Spectrosc. 2014, 45, 1059–1067. [Google Scholar] [CrossRef]
- Kaszewska, E.A.; Sylwestrzak, M.; Marczak, J.; Skrzeczanowski, W.; Iwanicka, M.; Szmit-Naud, E.; Anglos, D.; Targowski, P. Depth-Resolved Multilayer Pigment Identification in Paintings: Combined Use of Laser-Induced Breakdown Spectroscopy (LIBS) and Optical Coherence Tomography (OCT). Appl. Spectrosc. 2013, 67, 960–972. [Google Scholar] [CrossRef]
- Van Loon, A.; Noble, P.; de Man, D.; Alfeld, M.; Callewaert, T.; Van der Snickt, G.; Janssens, K.; Dik, J. The Role of Smalt in Complex Pigment Mixtures in Rembrandt’s Homer 1663: Combining MA-XRF Imaging, Microanalysis, Paint Reconstructions and OCT. Herit. Sci. 2020, 8, 90. [Google Scholar] [CrossRef]
- Adam, A.J.L.; Planken, P.C.M.; Meloni, S.; Dik, J. TeraHertz Imaging of Hidden Paint Layers on Canvas. Opt. Express 2009, 17, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Picollo, M. Terahertz Spectroscopy Applied to the Analysis of Artists’ Materials. Appl. Phys. A 2010, 100, 591–597. [Google Scholar] [CrossRef]
- Picollo, M.; Fukunaga, K.; Labaune, J. Obtaining Noninvasive Stratigraphic Details of Panel Paintings Using Terahertz Time Domain Spectroscopy Imaging System. J. Cult. Herit. 2015, 16, 73–80. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Backing | Sample Name | Measure 1 [µm] | Measure 2 [µm] | Measure 3 [µm] | Instrument Precision [µm] | Average Thickness [µm] | σ [µm] |
|---|---|---|---|---|---|---|---|
| OC-FIX17 | F4 | 56 | 68 | 57 | 1 | 60 | 7 |
| Acetate | F3 | 68 | 109 | 108 | 1 | 95 | 23 |
| Standard | A | 126 | 128 | 80 | 1 | 111 | 27 |
| Parafilm | C1 | 127 | 136 | 132 | 1 | 132 | 5 |
| Plastic | C1 | 156 | 184 | 182 | 1 | 174 | 16 |
| OC-FIX (Dis) | B3 | 210 | 204 | 200 | 1 | 205 | 5 |
| OC-FIX (Dis) | B4 | 296 | 216 | 204 | 1 | 239 | 50 |
| OC-FIX | AR1 | 306 | 318 | 329 | 1 | 318 | 12 |
| Fast Scanning | High-Resolution Scanning | |
|---|---|---|
| Time [s] | 389.1 | 1188.0 s |
| Number of pixels | 99 × 132 | 200 × 199 |
| Pixel dimensions [mm × mm] | 2 × 1.512 | 1 × 1 |
| Collimator diameter [mm] | 2 | 1 |
| Integration time [ms/pixel] | 30 | 30 |
| Current [mA] | 100 | 200 |
| Tension [kV] | 50 | 50 |
| Filtering, anode materials | No filter, Rh | No filter, Rh |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zito, R.; Bonizzoni, L.; Ludwig, N. Application of Macro X-ray Fluorescence Fast Mapping to Thickness Estimation of Layered Pigments. Sustainability 2024, 16, 2467. https://doi.org/10.3390/su16062467
Zito R, Bonizzoni L, Ludwig N. Application of Macro X-ray Fluorescence Fast Mapping to Thickness Estimation of Layered Pigments. Sustainability. 2024; 16(6):2467. https://doi.org/10.3390/su16062467
Chicago/Turabian StyleZito, Riccardo, Letizia Bonizzoni, and Nicola Ludwig. 2024. "Application of Macro X-ray Fluorescence Fast Mapping to Thickness Estimation of Layered Pigments" Sustainability 16, no. 6: 2467. https://doi.org/10.3390/su16062467
APA StyleZito, R., Bonizzoni, L., & Ludwig, N. (2024). Application of Macro X-ray Fluorescence Fast Mapping to Thickness Estimation of Layered Pigments. Sustainability, 16(6), 2467. https://doi.org/10.3390/su16062467