1. Introduction
To this day, arsenic remains one of the best known and most notorious poisons, thanks to numerous prominent historical assassinations [
1]. It is acutely toxic if swallowed or inhaled, it is carcinogenic, it is also toxic to aquatic organisms and therefore it is one of the 10 chemicals of greatest concern for human health [
2]. Trivalent arsenic shows the highest toxicity and mobility compared to pentavalent arsenic or organically bound arsenic. Consequently, arsenic is a toxin for humans and the environment, which is subject to strict regulations and limits in Europe [
3].
In many places, for example in Chile, the copper industry’s mines reach deeper layers of rock, which contain more sulphide minerals than oxide minerals and these same sulphide minerals come with higher amounts of arsenic. It is estimated that about 70% of the world’s copper reserves are in the form of chalcopyrite (CuFeS
2) [
4], which is arsenic-free and occur together with the minerals arsenopyrite (FeAsS) or enargite (Cu
3AsS
4), which contain arsenic as a main component. However, conventional flotation separation does not separate these minerals from each other, but only from copper-free gangue. The resulting copper concentrate therefore still contains the arsenic, which then enters the further copper-extraction process [
5].
As copper is an extremely important technological metal and is found in almost all electronic devices, it is an irreplaceable part of our current industry and everyday life. The increasing arsenic content in copper ore, combined with the toxic properties of arsenic, is thus an issue that needs to be addressed [
6].
Since the arsenic cannot be separated in the existing flotation system, it enters the smelting furnaces with the copper concentrate [
5]. However, arsenic passes into the gas phase at comparatively low temperatures and is therefore subsequently collected in a gas scrubber in sulphuric acid solution. Alternatively, the arsenic can be separated in a roasting process before the furnaces, but then also ends up in a gas scrubber. For landfilling, the arsenic must be precipitated from this solution. The current state of the art is to precipitate the trivalent arsenic as calcium arsenite (AsCaHO
3). Unfortunately, this compound is easily soluble in water and hence difficult to landfill without the risk of emitting arsenic. Alternatively, arsenic(III) is xidized to arsenic(V) and then precipitated as a calcium or iron compound. The latter, also called scorodite (Fe
3+[AsO
4]·2H
2O), is a much more stable form and thus better qualified for landfilling [
7,
8].
The required oxidation Is usually realised with hydrogen peroxide (H
2O
2), but as this is an expensive method, oxidation is often completely dropped for cost reasons. There are various alternative oxidation options, which have been extensively investigated in recent years. One example of these is photocatalytically driven oxidations. The irradiation of water with UV light alone leads to the production of hydroxyl radicals, which in turn oxidise the arsenic. Titanium oxide, for example, can be used as a supporting catalyst [
9]. Photoelectrochemical oxidation also works with sunlight. Here, semiconductor electrodes are used to achieve the oxidation [
10]. One of the main problems with both methods is the provision of sufficient UV or sunlight. The aforementioned studies oxidise arsenic in the mg/L range, while the As(III) concentration in the effluent of a roaster used in Chile, for example, is in the range of 18 g/L. A more exotic oxidation method is sonochemical oxidation—oxidation by ultrasonic waves. This can still be made more efficient by using additives, such as peroxodisulfate (PDS) [
11]. Again, oxidation of arsenic in the low mg/L range is being studied. Providing sufficiently large ultrasonic tanks and the required amount of PDS for the conditions targeted in this work (18 g/L As(III); ~2000 m
3/d) appears uneconomical. More common oxidation methods include the use of ozone as an oxidant or catalytic support of hydrogen peroxidation using iron ions (Fenton reaction) [
12,
13]. However, proof of feasibility and usefulness in the targeted conditions has yet to be provided. This work focuses on the new approach of electrochemical oxidation of As(III) to As(V) using boron-doped diamond (BDD) electrodes and mainly investigates the feasibility in extremely high arsenic concentrations and compares the viability with the state of the art oxidation using hydrogen peroxide.
2. Experimental Section
The most important part of the experimental setup is the boron-doped diamond electrode. For this work, the electrode was purchased from the company DiaCCon. The quality of the electrodes on the market differs significantly in the robustness and durability of the electrodes. For this type of electrode, a carrier material, in our case niobium, is coated with a diamond layer. The diamond layer is ≥12 µm thick and doped with boron to achieve conductivity [
14]. Stainless steel was used as the cathode material.
At the beginning of our investigations, As2O3 was dissolved in sodium hydroxide solution to prepare the arsenic solution (alkaline route). For this purpose, the arsenic oxide was first dissolved in a 7.5% sodium hydroxide (NaOH) solution at 40 °C and the liquor was then diluted again 1:1 with water; the final concentration was therefore 3.75% NaOH. This procedure succeeded without problems up to a concentration of 20 g/L of arsenic. The typical concentration of arsenic in the gas-scrubber solution of a roaster used in Chile is about 18 g/L. In the experiments, an orientation was attempted to this very high arsenic concentration. In order to simulate the conditions on site more realistically, at a certain point the arsenic was dissolved in sulphuric acid (acidic route). For this purpose, the weighed arsenic oxide was ground to a slurry in a mortar with concentrated sulphuric acid and then added in portions to a 2-molar sulphuric acid at 85 °C. The solution was stirred for a further 3 h at the same temperature and then left to cool overnight. However, precipitation occurred while cooling and over a longer standing time, and was subsequently filtered off. All chemicals used were in p.a. purity.
The standard experiment was carried out with a 40 cm
2 electrode area. The arsenic solution to bexidizedd was continuously mixed during the experiments by means of a magnetic stirrer. The electrode distance between the BDD electrode (anode) and steel electrode (cathode) was always 5 mm. The current used was 2 A, and thus 50 mA/cm
2. The electrochemical oxidation always started at room temperature.
Figure 1 shows the experimental setup.
To monitor the oxidation progress, samples were taken regularly during the experiment and directly at its end. The analytical differentiation of arsenic(III) and arsenic(V) was carried out photometrically via the formation of molybdenum blue in the presence of arsenic(V). For this purpose, a ready-made rapid test determination for phosphate in water samples offered by the company Hach Lange GmbH (Berlin, Germany) was transferred to the purpose of arsenic differentiation [
15]. The total arsenic content was determined by means of the Optima 8300 from PerkinElmer (Waltham, MA, USA), an optical emission spectrometer with inductively coupled plasma (ICP-OES).
3. Results
3.1. Previous Results
As already reported in the conference paper for COPPER 2022 [
16], proof of concept was achieved with 200 mL of an arsenic solution (760 mg/L As(III)) of the alkaline route with both 1 A and 2 A, respectively, using a 40 cm² BDD electrode. After only 15 min for 1 A and after 10 min for 2 A, only pentavalent arsenic could be detected in the solution.
Oxidation by means of a BDD electrode does not take place directly at the anode. Instead, indirect oxidation takes place. Hydroxyl radicals are formed in the water, which in turn oxidise the arsenic(III) to arsenic(V) [
17].
Therefore, Faraday’s law can theoretically be applied only to the formation of hydroxyl radicals and not to calculations of the oxidation of As(III) to As(V). Assuming that each hydroxyl radical carries with it an oxidizing power of one “z = chemical charge number”, at least the linear relationship remains.
In experiments, also published in the conference paper [
16], it could be shown that the real oxidation time is longer than the one calculated via the Faraday’s law, especially for low concentrations. At high arsenic concentrations (up to 20 g/L) the experimentally detected time becomes more and more similar to the calculated time. This suggests that the indirect character of the oxidation has an influence on the time, which is greater the lower the arsenic concentration. The Faraday law can therefore be used as an approximation at high arsenic concentrations with the help of a correction factor which reflects the efficiency of the indirect oxidation.
n = amount of substance (mol);
m = mass (g);
M = molar mass (g/mol);
I = current (A);
t = time (s);
z = chemical charge number;
F = Faraday constant (C/mol = A * s/mol).
There are other influencing factors which do not appear in Faraday’s formula. For example, the deviation between theoretical and actual oxidation time is particularly large at low arsenic concentrations. In the corresponding experiments (130 mL; 2 A; 40 cm² electrode area, alkaline path) it was found that at a concentration of 1 g/L As(III) the real oxidation time was five times longer than the calculated one, while at 20 g/L the deviation from theory was only 10% [
16]. The reason for this was the diffusion effects which prevented an optimal oxidation process.
The electrode area does not appear in Faraday’s formula, although it must also be taken into account. Thus, a comparison of two experiments with different electrode sizes shows that the oxidation time increased with decreasing electrode size. For example, the entire oxidation of an arsenic solution containing 7.4 g/L As(III) (100 mL; 2 A) took 84 min with a diamond electrode of only 1 cm², while the same solution under the same conditions was completelyxidizedd after only 19 min with an electrode of 40 cm². At the same time, a noticeably higher resistance was measured with the smaller electrode, which gives an explanation for the poorer efficiency. The manufacturer of the electrodes specifies a current density of 100 mA/cm² as the optimum current intensity [
14].
We have shown, in a series of experiments, that steel electrodes as the cathode show the same performance for oxidation as graphite or another BDD electrode [
16]. Apart from this one series of experiments, steel was always chosen as the cathode in addition to the BDD anode in all other experiments. Most of the literature on BDD electrode oxidation, such as that of Panizza and Martinez-Huitle [
19], gives little or no consideration to the cathode, and although only rudimentary tests have been carried out here, this is certainly a topic that needs further investigation, to alsoxidizede the cathode reaction. This reaction depends on the side ions which are present in the solution, for example, Cu
2+ is reduced at the cathode if it is present in the solution. However, the cathode reactions were not investigated in this work.
3.2. Side Ions in Solution
In the real water of a gas scrubber, the solution is not alkaline, as in the experiments presented in the previous chapter (or in more detail in the mentioned conference paper [
16]), but consists of sulphuric acid. In addition, other ions are present in the solution, which could cause side reactions. Since real waste water was hardly available, model solutions were used.
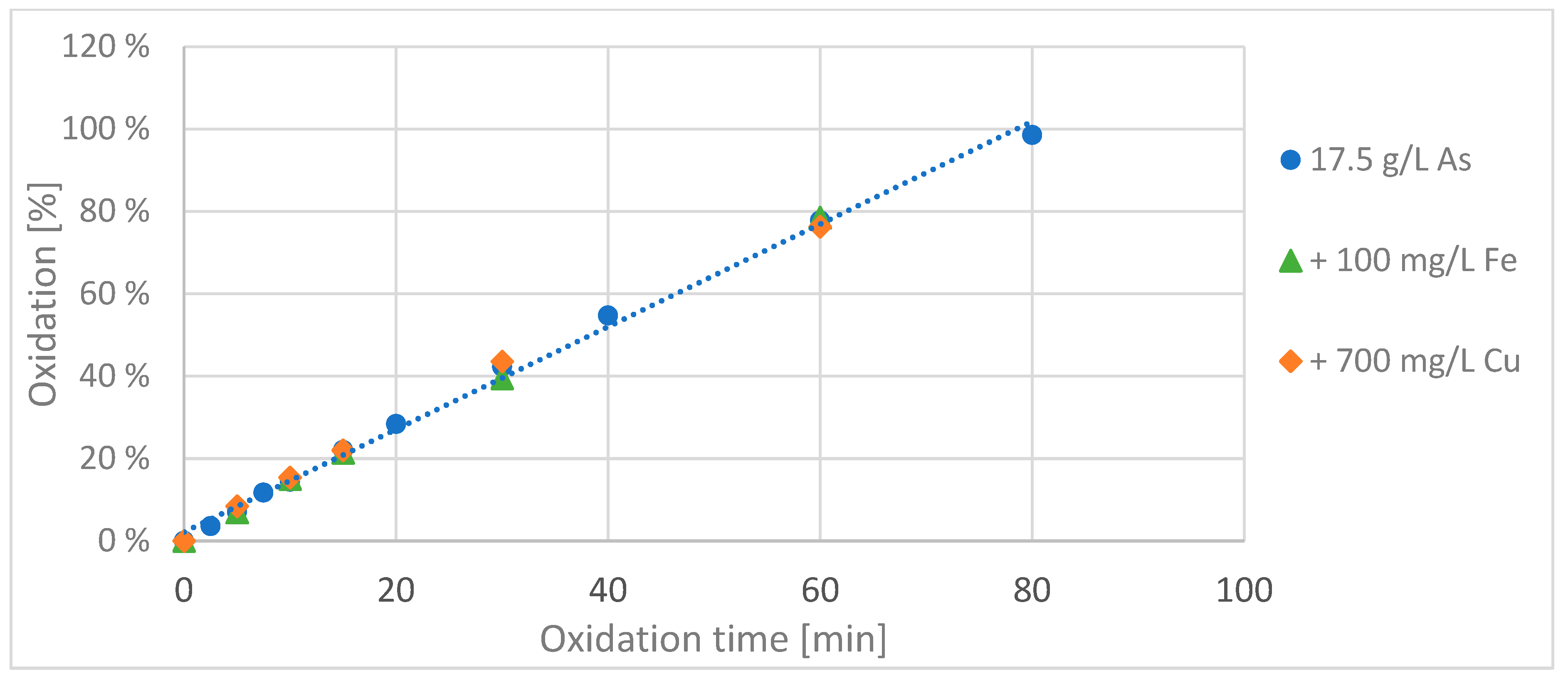
In the first series of experiments, iron was added to the arsenic solution in one batch, copper was added to another batch, and in the third batch only the pure arsenic solution wasxidizedd using a BDD electrode. The oxidation course of 200 mL of the two resulting solutions with additional ions were compared with the oxidation course of the same volume of a pure arsenic solution (see
Figure 2).
The oxidation of the solutions with the side ions was monitored only for the first 60 min. It can be clearly seen that in this time there was no influence of the interfering ions on the oxidation rate of arsenic(III) to arsenic(V). During the oxidation process the iron concentration remained the same over the entire oxidation time, while the copper concentration decreased linearly in the first 30 min, dropping from 700 mg/L to about 100 mg/L, due to a reduction of the copper at the cathode. In the remaining 30 min of the experiment, the copper concentration in solution reduced further, but more slowly, to a value of about 60 mg/L.
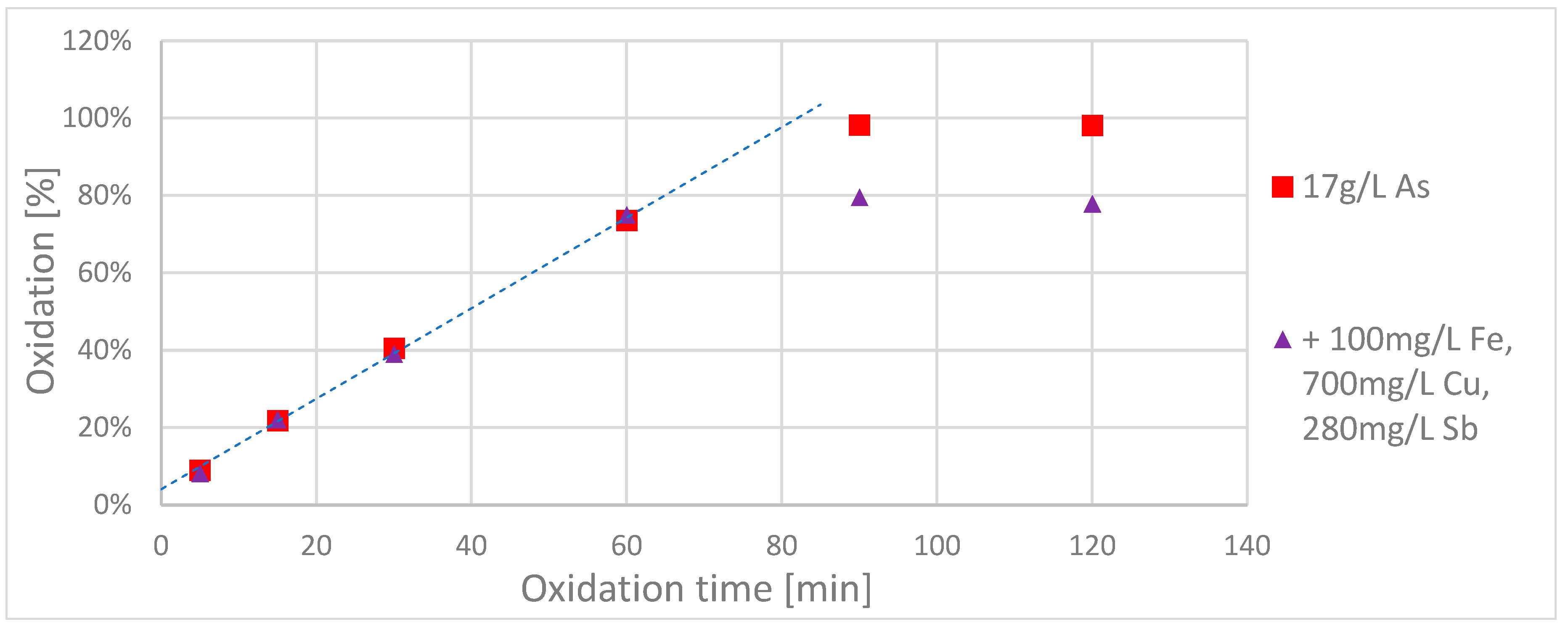
To clarify whether different ions in combination have a negative influence on the oxidation process, a sulphuric acid solution was prepared that contained ~17 g/L arsenic(III), 0.1 g/L Fe(II), 0.7 g/L Cu(II) and 0.28 g/L Sb(III) in one batch and 200 mL of this solution wasxidizedd with 40 cm² BDD electrode area and a current of 2 A (acidic route).
This time the three interfering ions mentioned (Fe, Cu, Sb) were added simultaneously. The oxidation course was again compared with that of 200 mL of a pure arsenic solution, see
Figure 3.
In contrast to the previous experiment, the arsenic concentration was monitored longer for both solutions. As before, the interfering ions had no relevant influence on the oxidation rate for the first 60 min. Surprisingly, however, the oxidation rate of the solution with the additional ions seemed to stagnate after 60 min at a level of about 80%, while the pure arsenic solutionxidizedd up to 100%.
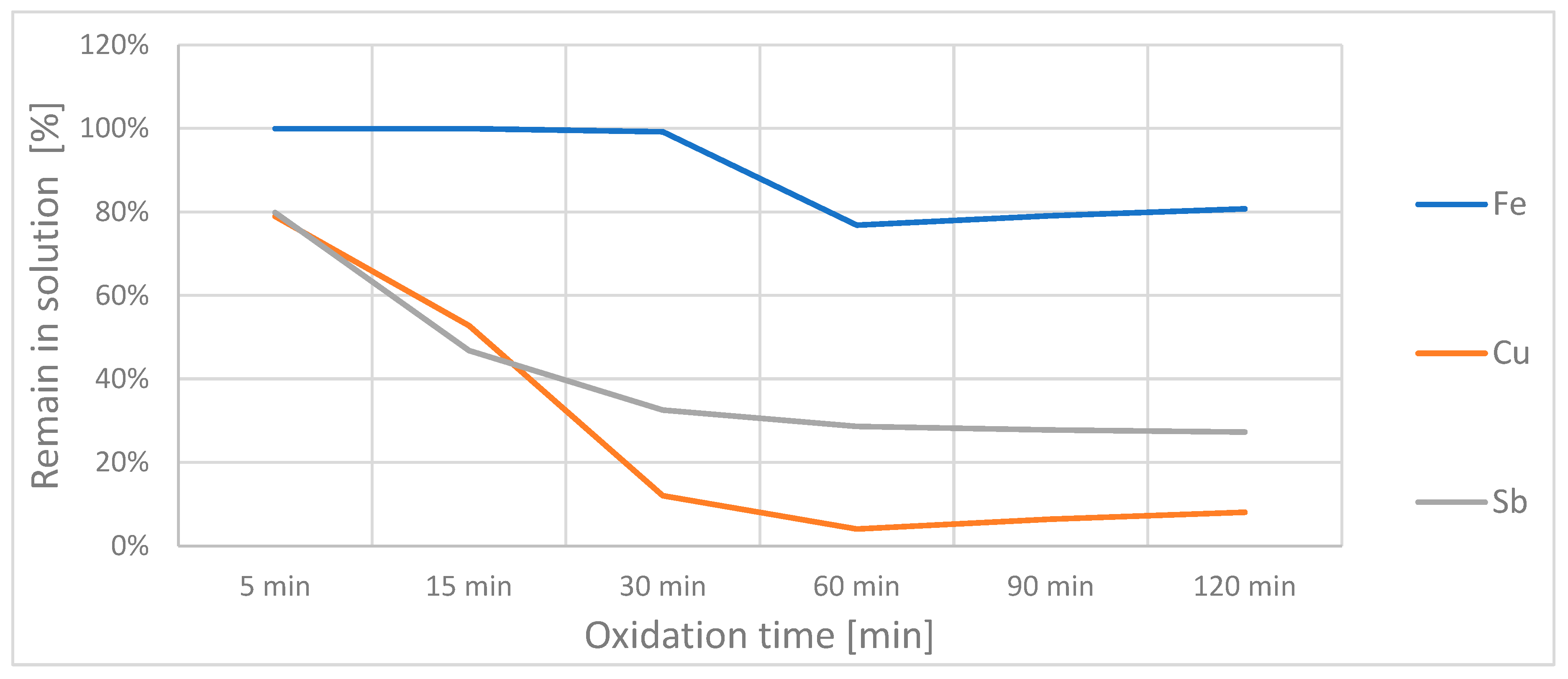
The change in concentration of the additional ions in solution during oxidation can be seen in
Figure 4. The iron concentration again remained relatively constant, although it decreased slightly between 30 and 60 min; the copper and antimony concentrations decreased steadily until they stagnated at a level of ~5% and ~30%, respectively.
One explanation for the stagnation of the oxidation after 60 min could be a passivating layer that formed on the steel cathode during the oxidation test. This was a brown-black muddy residue. A semiquantitative analysis via ICP-OES resulted in a weight ratio As:Cu:Sb:Fe of 4000:1000:50:1. The residue basically contained arsenic, copper and some antimony.
Of the original arsenic(III), 80% wasxidizedd to As(V) and about 10% could no longer be found in the solution after the experiment and presumably migrated into the residue on the cathode.
This effect could not be seen in the first experiment in
Figure 2 because the two solutions with interfering ions were only monitored for 60 min, and hence the effect had not yet occurred.
Before upscaling the use of these electrodes to a large technical application, it is essential further to investigate this effect of the passivating layer formed by the side ions.
3.3. Precipitation of Scorodite
Since the oxidation of arsenic is necessary to precipitate it in the very stable form of scorodite, the latter was precipitated in the course of this work.
To obtain an arsenic-binding solid, which is as insoluble as possible, it is important that the scorodite precipitates in a very crystalline form. To achieve this, comparatively high temperatures of about 90 °C, as well as low pH of about 1 are needed. In addition, precipitation has to be initiated by seed crystals. A precipitation solely via a pH shift would result in amorphous material [
20,
21].
Since scorodite is not easily obtainable as a pure chemical we relied on natural crystals as seed crystals (
Figure 5 left). The crystals were removed from the rock as pure as possible and then ground in a mortar (
Figure 5 right).
As a starting solution, 250 mL of an As(III) solution was prepared according to the acidic route andxidizedd electrochemically with the BDD electrode (40 cm², 2 A) for 180 min. The arsenic concentration was supposed to be adjusted to 20 g/L, but since precipitation occurred during the overnight cooling, the solution had only 15 g/L As(III). This was taken into account, but did not influence the significance of the experiment. About 95% of this remaining arsenic could bexidizedd to As(V), so that the resulting initial solution for the precipitation experiment had an As(V) concentration of 14.3 g/L.
In the next step, iron was added in the form of ferric sulphate. The amount of ferrous sulphate was calculated molar equivalent to 14.3 g/L arsenic, as Singhania et al. showed that an equivalent addition of iron is sufficient [
20]. The ferric sulphate dissolved rapidly within 15 min in the highly acidic solution.
At a temperature of just under 100 °C, the pH was raised with NaOH until the white flakes that briefly formed when the base was added no longer dissolved on their own (pH = 1.1). Then sulphuric acid was added again until the solution was (almost) clear again (pH = 0.87, after the procedure described in Singhania et al. [
20]).
At this point, the seed crystals (1.1 g ground natural scorodite crystals) were added. The precipitation did not proceed instantly suddenly afterwards, but successively (See
Figure 6).
After 150 min of precipitation time, the experiment was stopped and the solution was filtered. The weight of the solid after drying was 5.97 g. Theory reveals thatxidizex. 21 mmol arsenic and 20 mmol iron must have been removed from the solution. Based on the iron, this could result in 4.6 g of scorodite. Adding the 1.1 g of seed crystals resulted in a balance of 5.7 g. Possible explanations for the missing 0.3 g could be other arsenic compounds and small measurement error in the concentration determination.
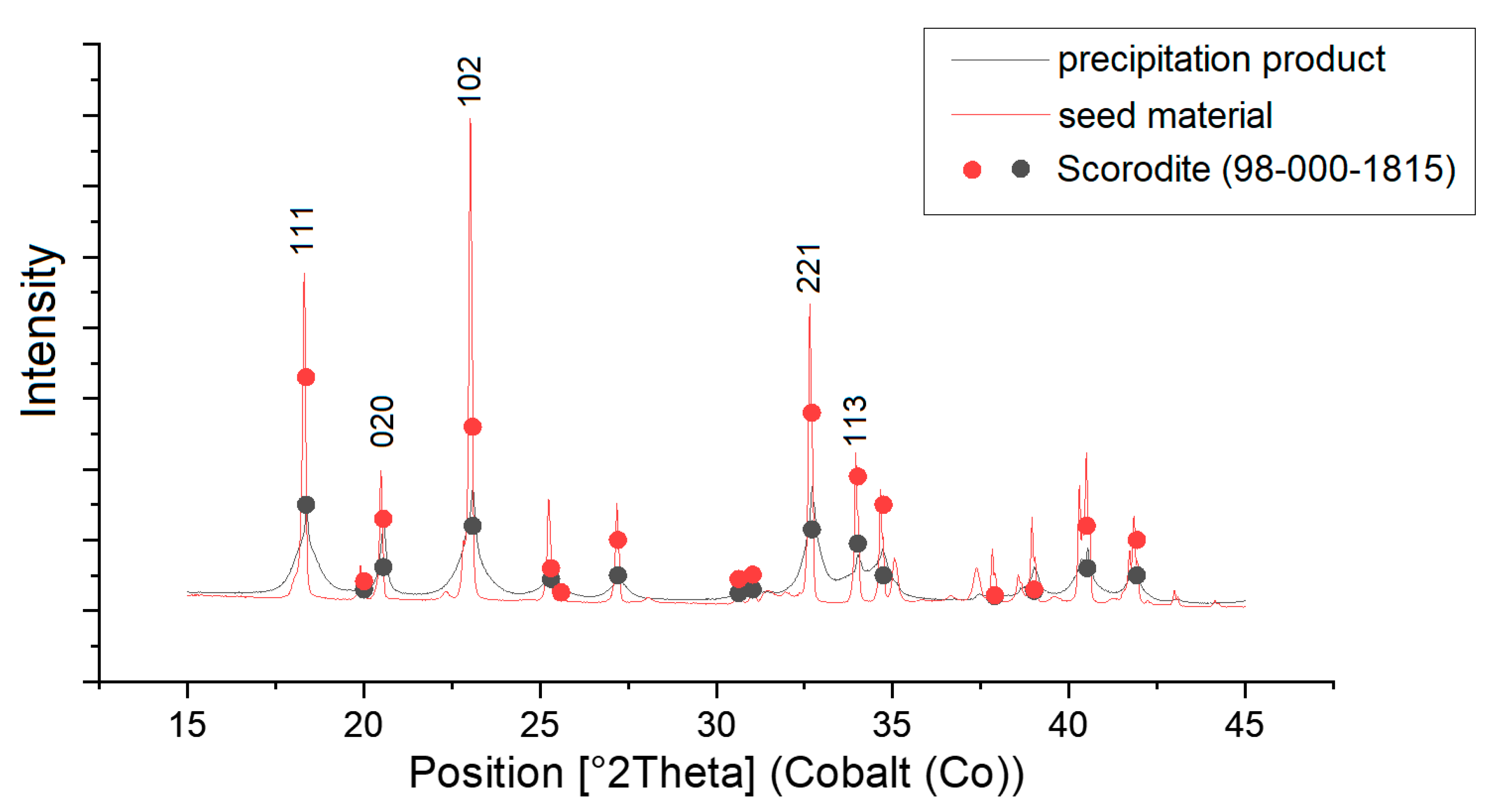
To verify that the precipitated product was scorodite, XRD was used to compare the seed material and the precipitated product with data from the database (see
Figure 7).
It is easy to see that both the naturally grown scorodite, which was used as a seed crystal, and the precipitated product fit very well with the markers of the scorodite database. The peaks of the natural crystals are slightly sharper than those of the precipitated product, which indicates a better crystallinity. Nevertheless, it can be seen that the precipitate is also predominantly crystalline [
22].
Crystallization of the entire arsenic in solution would have taken 6 h at a constant crystallization rate. According to Singhania et al., the precipitation rate is positively influenced by a larger amount of seed crystals [
20]. In the experiment only a small amount could be used due to a lack of crystalline scorodite. In a large-scale continuous process, it is feasible to provide a large quantity of seed crystals, and hence reduce the crystallization time.
4. Economic Efficiency Analysis
In order to get an idea of the costs and to compare them with oxidation via hydrogen peroxide, the OPEX costs for the oxidation of one ton of arsenic for both oxidation methods (BDD and H2O2) are first estimated.
In
Figure 2 and
Figure 3 it can be seen clearly that in a pure arsenic solution for 200 mL of a 17.5 g/L (
Figure 2) and a 17 g/L (
Figure 3) it took 80 min to oxidise 100% of As(III) to As(V) with 2 A, respectively. After solving Faraday’s law for the time, one obtains 75 and 73 min, respectively, for these two solutions, in an ideal process.
n = amount of substance (mol);
m = mass (g);
M = molar mass (g/mol);
V = volume (L);
c = concentration (g/L);
I = current (A);
t = time (s);
z = chemical charge number;
F = Faraday constant (C/mol = A * s/mol).
Thus, the efficiency of electrochemical oxidation was about 94% and 91%, respectively. For the further calculation, we assume an average efficiency of 92.5%.
One tonne of arsenic corresponds to 13,350 moles of As rounded, which according to Faraday require just under 540 kA for complete oxidation in 80 min.
Because of the efficiency losses in a real process, one would need 580 kA.
Assuming a voltage of 5 V, which was the average voltage achieved in the experiments, this results in 2900 kW and through the 80 min processing, in about 3870 kWh. According to GlobalPetrolPrices, the kilowatt hour costs USD 0.113 for industry in Chile in October 2022. This results in an OPEX price for the oxidation of one ton of arsenic with BDD electrodes of approximately USD 440 [
23].
Currently, arsenic isxidizedd by means of hydrogen peroxide. The reaction proceeds stoichiometrically in a molar ratio of 1:1.
In practice it is common to work with a surplus. For our calculation we assume a surplus of 50%, so 20,000 moles of H
2O
2 are needed. With a molar mass of 34.02 g/mol for H
2O
2, this gives 0.7 tons of peroxide. According to Ecoinvent, 1 ton of 50% hydrogen peroxide costs about USD 460 on the global market, excluding transport [
24]. The amount of hydrogen peroxide (100%) needed to oxidise one ton of arsenic therefore costs USD 640.
In the comparison of both methods, additional transport would be incurred for the hydrogen peroxide, which must be taken into account in the calculation. A 20 standard container has a maximum cargo weight of about 28 tons [
25]. The transport of such a container is estimated at about USD 3900 in the following calculation [
26]. Thus, the transport costs about USD 140 per ton H
2O
2.
For the BDD electrode, on the other hand, the acquisition costs of the electrode material must be considered, assuming that the other structural KPEX costs will be approximately the same for both methods.
The current density recommended by the manufacturer during the use of the electrode is 100 mA/cm² [
14]. In the consideration made above, 580 kA is required, which then requires an electrode area of 580 m². With a utilization rate of 90% and the calculated electrode size, about 5900 tons of arsenic could be converted per year.
The lifetime of a BDD electrode has not been investigated within this work. It can be assumed that the voltage level has an influence on the lifetime and therefore has to be controlled [
27]. Additionally, the presence of organics, which form alkyl radicals during oxidation, appears to drive corrosion of the electrode [
28]. In our experiments, at 5 V, the voltage was moderate compared to a voltage of 10 V which might lead to significant decomposition of the BDD layer [
27]. Furthermore, no organics are expected in the arsenic solution of the gas treatment. Concerning a degradation in strong acidic solutions correspondence with the manufacturer revealed an observed lifetime of the electrodes in strong sulphuric acid of at least 10 years. Therefore, a lifetime of 10 years is a good assumption for further calculations.
The current market price for BDD electrodes from the manufacturer DiaCCon is about USD 18,800 per square meter [
14]. However, BDD electrodes are currently niche products, although through our correspondence with the manufacturer, it was determined that the price can be reduced drastically under very favourable assumptions, i.e., large-scale and on-site production. In that case the price could be decreased to approximately USD 6500–7000 per square metre of BDD electrode. For further calculations, a price per square metre of USD 7000 is therefore assumed. The required 580 m² would thus costxidizex. USD 4 million. Assuming use over 10 years and the quantity of arsenic converted per year calculated above, additional KPEX costs of USD 70 (rounded) per ton of arsenic must be added.
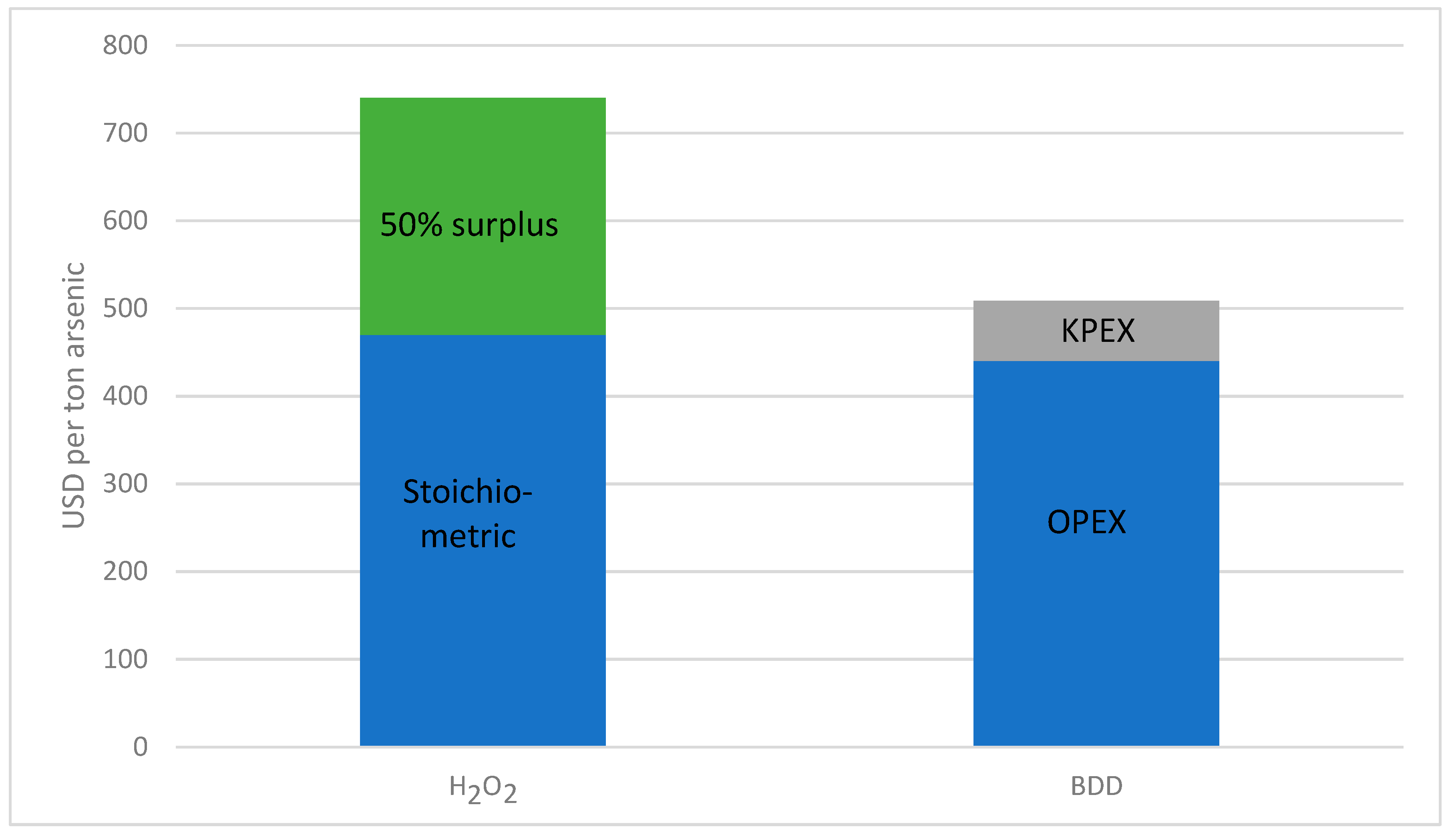
Figure 8 compares the cost of oxidizing one ton of arsenic with hydrogen peroxide or using electrochemical oxidation.
Over the calculated amount of 5900 t/a arsenic, about USD 1.4 million (rounded) could be saved annually by using a BDD electrode.
5. Conclusions
It can bexidizeddd that the oxidation of arsenic by means of a BDD electrode works reliably and follows a linear law. Even high concentrations of arsenic can bexidizedd from arsenic(III) to arsenic(V) within a relatively short time. For example, 200 mL of a solution containing 17.5 g/L arsenic was completelyxidizedd within 80 min.
Interfering ions in the solution can certainly complicate or negatively influence the electrochemical reaction. In low concentrations, it was shown that the oxidation rate was quite comparable to a pure arsenic solution, but that a layer was formed on the cathode, which contained both arsenic and other interfering ions. In the end, an oxidation of 100% could not be achieved, in contrast to the pure arsenic solution. A potential solution to this problem could be a continuous design in which the solution flows through different electrode sections or oxidation tanks after different oxidation times, so that deposition on the cathode can take place more selectively. In addition, a different choice of cathode material could also influence the results. Further research is needed here.
As expected, the electrochemicallyxidizedd arsenic(V) can be precipitated as scorodite with the addition of iron salts and seed crystals. The conditions must be controlled precisely (slightly under 100 °C; pH~1; seed crystals) for a crystalline product to precipitate. The feasibility could be demonstrated by a test precipitation with subsequent XRD analysis.
As calculated in the chapter on economic efficiency, electrochemical oxidation via BDD electrodes is the cheaper method compared to oxidation with hydrogen peroxide, and under the assumption of a large production, the investment costs would be reasonable. It could be calculated that for the oxidation of one ton of arsenic about 0.7 tons of hydrogen peroxide (100%; incl. 50% surplus) would be needed. The cost of this, including transportation, runs to about USD 740. Whereas the costs for oxidation of the same amount of arsenic by means of BDD electrodes, including the initial costs (lifetime 10 years), amount to only about USD 510.
Assuming lower electricity costs, e.g., through a solar park for self-use, the price advantage of electrochemical oxidation would probably be even higher. Another advantage is the resulting independency from the global hydrogen peroxide market.
As an overall conclusion, electrochemical oxidation of arsenic solutions from the copper production using BDD anodes is a very promising method with highly attractive economic advantages compared to the use of hydrogen peroxide.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}