
Acute Promyelocytic Leukemia in a Woman with Thalassemia Intermedia: Case Report and Review of Literature on Hematological Malignancies in β-Thalassemia Patients

 , , and
, , and
Abstract
1. Introduction
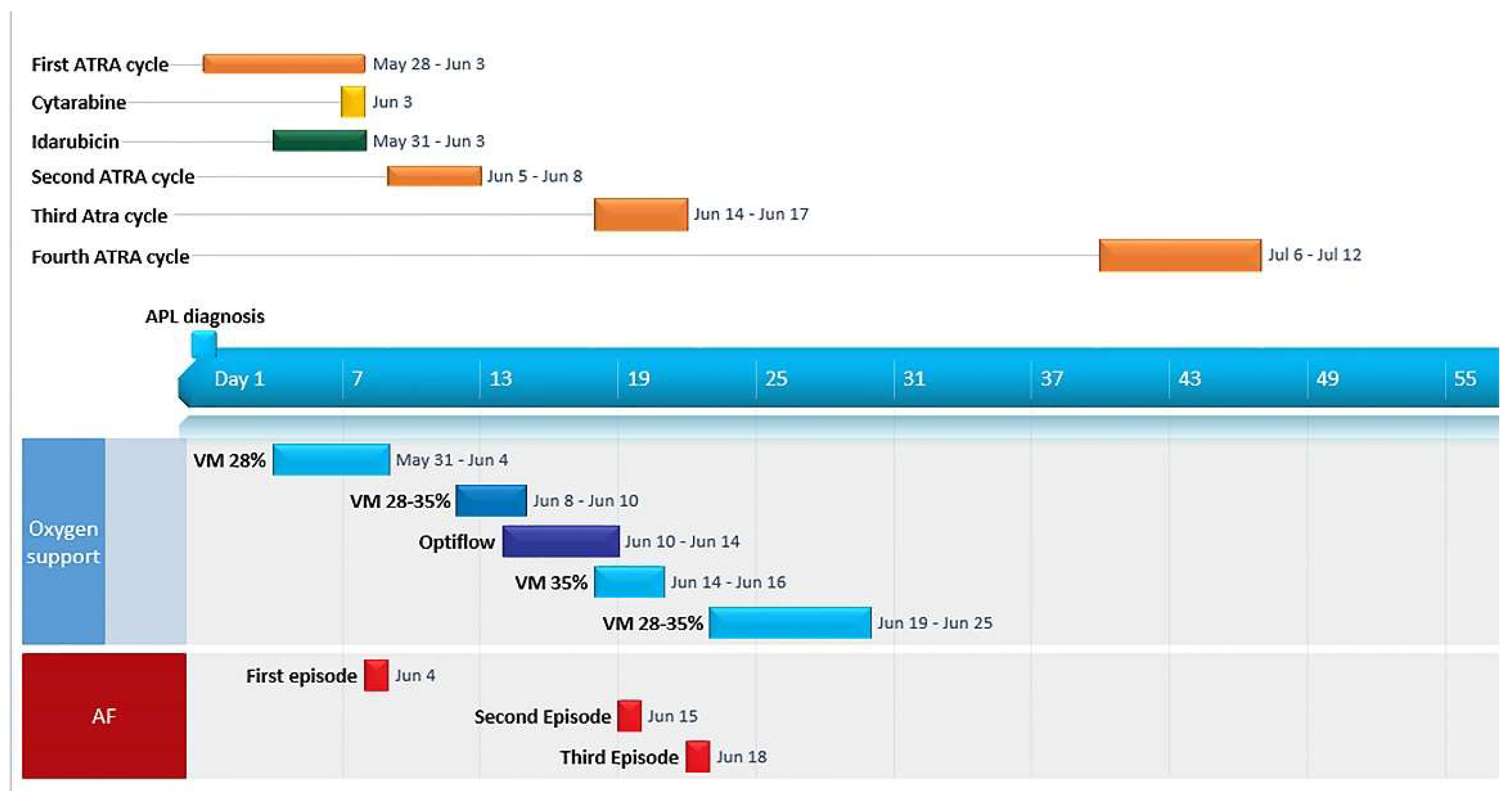
2. Case Report
3. Systematic Review of Studies on β-Thalassemia and Hematological Malignancies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Taher, A.T.; Cappellini, M.D. How I manage medical complications of b-thalassemia in adults. Blood 2018, 132, 1781–1791. Available online: http://ashpublications.org/blood/article-pdf/132/17/1781/1468099/blood818187.pdf (accessed on 18 May 2022). [CrossRef]
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellström-Lindberg, E. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717. Available online: https://haematologica.org/article/view/5076 (accessed on 18 May 2022). [CrossRef] [PubMed]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. Available online: https://pubmed.ncbi.nlm.nih.gov/30803991/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Breccia, M.; Gallo, E.; Rambaldi, A.; Paoloni, F.; Fioritoni, G.; Ferrara, F.; Specchia, G.; et al. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: Results of the AIDA-2000 trial of the GIMEMA Group. Blood 2010, 116, 3171–3179. [Google Scholar] [CrossRef]
- Balestrazzi, P.; Butturini, A. Thalassemia and Hodgkin lymphoma. Blut 1985, 50, 55. [Google Scholar] [CrossRef]
- Thapa, R.; Pal, P.K.; Mukhopadhyay, M. Beta Thalassemia Major and Hodgkin Lymphoma. J. Pediatr. Hematol. 2009, 31, 462–463. Available online: https://pubmed.ncbi.nlm.nih.gov/19648798/ (accessed on 18 May 2022). [CrossRef]
- Jabr, F.I.; Aoun, E.; Yassine, H.; Azar, C.; Taher, A. β-thalassemia intermedia and Hodgkin lymphoma. Am. J. Hematol. 2006, 81, 151. Available online: https://pubmed.ncbi.nlm.nih.gov/16432853/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Ahad Aziz Qureshi, P.A.; Razi, M.; Rehman, A.; Hassan, A. Hodgkin lymphoma with co-existing extramedullary haematopoiesis in a Thalasaemia Major patient: Killing two birds with one stone using PET-CT. J. Pak. Med. Assoc. 2019, 69, 1059. Available online: https://pubmed.ncbi.nlm.nih.gov/31983750/ (accessed on 18 May 2022).
- Tozzi-Cecchetti, M.C.; D’Amelio, R.; Baroni, C.D. Thalassemia major terminating in a non-Hodgkin B-cell lymphoma: Report of a case. Tumori 1979, 65, 353–357. [Google Scholar] [CrossRef]
- Schilirò, G.; Russo, A.; Marino, S.; Musumeci, S.; Russo, G. Occurrence of lymphoma with bone marrow involvement in a boy with beta+ thalassemia major. Clin. Lab. Haematol. 1979, 1, 325–328. Available online: https://pubmed.ncbi.nlm.nih.gov/544148/ (accessed on 18 May 2022). [CrossRef]
- Otrock, Z.K.; Shamseddine, A.I.; Taher, A.T. Non-Hodgkin disease in β-thalassemia major. Am. J. Hematol. 2005, 81, 62–64. Available online: https://pubmed.ncbi.nlm.nih.gov/16369971/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Ricchi, A.; Carta, A.; Pinna, G.M.; Cirio, E.M.; Foddanu, M.; Terrosu, P.F.; Pardini, S.; Fozza, C.; Longinotti, M. Asymptomatic cardiac lymphoma in a hepatitis C virus-positive thalassemic patient. Ital. Hear. J. 2004, 5, 302–304. Available online: https://pubmed.ncbi.nlm.nih.gov/15185891/ (accessed on 18 May 2022).
- Triantos, C.; Kourakli, A.; Kalafateli, M.; Giannakopoulou, D.; Koukias, N.; Thomopoulos, K.; Lampropoulou, P.; Bartzavali, C.; Fragopanagou, H.; Kagadis, G.C.; et al. Hepatitis C in patients with β-thalassemia major. A single-centre experience. Ann. Hematol. 2013, 92, 739–746. Available online: https://pubmed.ncbi.nlm.nih.gov/23412560/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Chehal, A.; Loutfi, R.; Taher, A. β-thalassemia intermedia and non-Hodgkin’s lymphoma. Hemoglobin 2002, 26, 219–225. Available online: https://pubmed.ncbi.nlm.nih.gov/12403486/ (accessed on 18 May 2022). [CrossRef]
- Tuğcu, D.; Karakaş, Z.; Gökçe, M.; Ağaoğlu, L.; Ünüvar, A.; Sarıbeyoğlu, E.; Akçay, A.; Devecioğlu, Ö. Thalassemia Intermedia and Acute Lymphoblastic Leukemia: Is it a Coincidental Double Diagnosis? Turk. J. Hematol. 2014, 31, 311. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4287036/ (accessed on 18 May 2022). [CrossRef]
- Kottaridis, P.D.; Peggs, K.; Lawrence, A.; Verfuerth, S.; Chatterjee, R.; Telfer, P.; Porter, J.B.; Mackinnon, S.; Goldstone, A.H. One antigen mismatched related donor bone marrow transplant in a patient with acute lymphoblastic leukemia and β-thalassemia major: Potential cure of both marrow disorders. Bone Marrow Transplant. 2000, 25, 677–678. Available online: https://pubmed.ncbi.nlm.nih.gov/10734304/ (accessed on 18 May 2022). [CrossRef][Green Version]
- Goyal, M.; Dinaker, M.; Gayathri, K. Adult T-cell leukemia/lymphoma and acquired immunodeficiency syndrome - CD4+ T-cell malignancy in CD4+ T-cell deficient status: A paradox. Indian J. Pathol. Microbiol. 2018, 61, 553–556. Available online: https://pubmed.ncbi.nlm.nih.gov/30303147/ (accessed on 18 May 2022). [CrossRef]
- Sherief, L.M.; Kamal, N.M.; Abdelrahman, H.M.; Hassan, B.A.; Zakaria, M.M. First report of acute lymphoblastic leukemia in an egyptian child with β-thalassemia major. Hemoglobin 2015, 39, 127–129. Available online: https://pubmed.ncbi.nlm.nih.gov/25707677/ (accessed on 18 May 2022). [CrossRef]
- Palomo-Colli, M.A.; Zapata-Tarres, M.; Castelán-Martínez, O.D.; Juárez-Villegas, L.E.; Córdova-Hurtado, L.P. A strategy for the clinical remission of acute lymphoblastic leukemia elicited by treatment of β-thalassemia major: A case report. Mol. Clin. Oncol. 2018, 8, 375–377. Available online: https://pubmed.ncbi.nlm.nih.gov/29435306/ (accessed on 18 May 2022). [CrossRef]
- Felici, W.; Ballati, G.; Vignetti, M.; Aliquò, C.; De Santis, F.; Tucciarone, L. Acute myeloid leukemia in a child affected by beta-thalassemia major. Am. J. Hematol. 1988, 29, 124. Available online: https://pubmed.ncbi.nlm.nih.gov/3189302/ (accessed on 18 May 2022). [CrossRef]
- Romani, C.; Murru, R.; Pettinau, M.; Origa, R.; Deplano, S.; Angelucci, E. Sudden cardiac failure in a β-thalassemia major patient receiving chemotherapy for acute promyelocytic leukemia. Leuk. Lymphoma 2007, 48, 628–629. Available online: https://pubmed.ncbi.nlm.nih.gov/17454611/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- De Soccio, G.; Savastano, V.; Minasi, S.; Bertin, S.; Serafini, M.; Vittori, T.; Riminuccie, M.; Corsi, A. Solitary juvenile xanthogranuloma of the hypopharynx. Clinico-pathologic study in a child with β-Thalassemia Major and Cutaneous Mastocytosis. Int. J. Pediatr. Otorhinolaryngol. 2020, 135, 110088. Available online: https://pubmed.ncbi.nlm.nih.gov/32505931/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Arora, S.; Ganapathi, K.A. Prominent pseudo-Gaucher cells in a patient with b-thalassemia intermedia and plasma cell myeloma. Blood 2020, 136, 2839. Available online: https://pubmed.ncbi.nlm.nih.gov/33301039/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Voskaridou, E.; Terpos, E.; Komninaka, V.; Eftyhiadis, E.; Mantzourani, M.; Loukopoulos, D. Chronic myeloid leukemia with marked thrombocytosis in a patient with thalassemia major: Complete haematological remission under the combination of hydroxyurea and anagrelide. Br. J. Haematol. 2002, 116, 155–157. Available online: https://pubmed.ncbi.nlm.nih.gov/11841409/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Wu, C.; Zhang, X.; Ye, X.; Chen, D.; Jin, J.; Huang, J. Co-existence of myeloproliferative neoplasias and β-thalassemia with IVS-2-654 mutation—a case report. Transl. Cancer Res. 2020, 9, 2069–2073. Available online: https://tcr.amegroups.com/article/view/36624/html (accessed on 18 May 2022). [CrossRef] [PubMed]
- Alavi, S.; Safari, A.; Sadeghi, E.; Amiri, S. Hematological malignancies complicating β-thalassemia syndromes: A single center experience. Blood Res. 2013, 48, 149–151. Available online: https://synapse.koreamed.org/articles/1092065 (accessed on 18 May 2022). [CrossRef]
- Benetatos, L.; Alymara, V.; Vassou, A.; Bourantas, K.L. Malignancies in β-thalassemia patients: A single-center experience and a concise review of the literature. Int. J. Lab. Hematol. 2008, 30, 167–172. Available online: https://pubmed.ncbi.nlm.nih.gov/18333849/ (accessed on 18 May 2022). [CrossRef]
- Karimi, M.; Giti, R.; Haghpanah, S.; Azarkeivan, A.; Hoofar, H.; Eslami, M. Malignancies in patients with β-thalassemia major and β-thalassemia intermedia: A multicenter study in Iran. Pediatr. Blood Cancer 2009, 53, 1064–1067. Available online: https://pubmed.ncbi.nlm.nih.gov/19533641/ (accessed on 18 May 2022). [CrossRef]
- Zurlo, M.G.; De Stefano, P.; Borgna-Pignatti, C.; Di Palma, A.; Piga, A.; Melevendi, C.; di Gregorio, F.; Burattini, M.; Terzoli, S. Survival and causes of death in thalassemia major. Lancet 1989, 2, 27–30. Available online: https://pubmed.ncbi.nlm.nih.gov/2567801/ (accessed on 18 May 2022). [CrossRef]
- Zanella, S.; Garani, M.C.; Borgna-Pignatti, C.; And, M.C.G. Malignancies and thalassemia: A review of the literature. Ann. N. Y. Acad. Sci. 2016, 1368, 140–148. Available online: https://pubmed.ncbi.nlm.nih.gov/26916208/ (accessed on 18 May 2022). [CrossRef]
- Chung, W.S.; Lin, C.L.; Lin, C.L.; Kao, C.H. Thalassemia and risk of cancer: A population-based cohort study. J. Epidemiol. Community Health 2015, 69, 1066–1070. Available online: https://pubmed.ncbi.nlm.nih.gov/25922472/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Montesinos, P.; Bergua, J.M.; Vellenga, E.; Rayón, C.; Parody, R.; De La Serna, J.; León, A.; Esteve, J.; Milone, G.; Debén, G.; et al. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: Characteristics, outcome, and prognostic factors. Blood 2009, 113, 775–783. Available online: https://pubmed.ncbi.nlm.nih.gov/18945964/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Leblebjian, H.; DeAngelo, D.J.; Skirvin, J.A.; Stone, R.M.; Wadleigh, M.; Werner, L.; Neubergae, D.S.; Bartelac, S.; McDonnellc, A.M. Predictive factors for all-trans retinoic acid-related differentiation syndrome in patients with acute promyelocytic leukemia. Leuk. Res. 2013, 37, 747–751. Available online: https://pubmed.ncbi.nlm.nih.gov/23643326/ (accessed on 18 May 2022). [CrossRef]
- Stahl, M.; Tallman, M.S. Differentiation syndrome in acute promyelocytic leukemia. Br. J. Haematol. 2019, 187, 157–162. Available online: https://pubmed.ncbi.nlm.nih.gov/31410848/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. Available online: http://www.nejm.org/doi/10.1056/NEJMra2021838 (accessed on 18 May 2022). [CrossRef]
- Eldor, A.; Rachmilewitz, E.A. The hypercoagulable state in thalassemia. Blood 2002, 99, 36–43. Available online: http://ashpublications.org/blood/article-pdf/99/1/36/1679491/h80102000036.pdf (accessed on 18 May 2022). [CrossRef]
- Cappellini, M.D.; Poggiali, E.; Taher, A.; Musallam, K. Hypercoagulability in β-thalassemia: A status quo. Expert Rev. Hematol. 2012, 5, 505–512. Available online: https://pubmed.ncbi.nlm.nih.gov/23146054/ (accessed on 18 May 2022). [CrossRef]
- Maia de Oliveira da Silva, J.P.; Brugnerotto, A.F.; Romanello, K.S.; Teixeira, K.K.L.; Lanaro, C.; Duarte, A.S.; Costa, G.G.L.; Araújo, A.S.; Bezerra, M.A.C.; Domingos, I.F.; et al. Global gene expression reveals an increase of HMGB1 and APEX1 proteins and their involvement in oxidative stress, apoptosis and inflammation pathways among beta-thalassemia intermedia and major phenotypes. Br. J. Haematol. 2019, 186, 608–619. Available online: https://pubmed.ncbi.nlm.nih.gov/31218684/ (accessed on 18 May 2022). [CrossRef]
- Kanavaki, I.; Makrythanasis, P.; Lazaropoulou, C.; Tsironi, M.; Kattamis, A.; Rombos, I.; Papassotiriou, I. Soluble endothelial adhesion molecules and inflammation markers in patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2009, 43, 230–234. Available online: https://pubmed.ncbi.nlm.nih.gov/19654073/ (accessed on 18 May 2022). [CrossRef]
- Butthep, P.; Rummavas, S.; Wisedpanichkij, R.; Jindadamrongwech, S.; Fucharoen, S.; Bunyaratvej, A. Increased circulating activated endothelial cells, vascular endothelial growth factor, and tumor necrosis factor in thalassemia. Am. J. Hematol. 2002, 70, 100–106. Available online: https://pubmed.ncbi.nlm.nih.gov/12111782/ (accessed on 18 May 2022). [CrossRef]
- Tang, L.; Chai, W.; Ye, F.; Yu, Y.; Cao, L.; Yang, M.; Xie, M.; Yang, L. HMGB1 promotes differentiation syndrome by inducing hyperinflammation via MEK/ERK signaling in acute promyelocytic leukemia cells. Oncotarget 2017, 8, 27314–27327. Available online: https://doi.org/10.18632/oncotarget.15432 (accessed on 18 May 2022). [CrossRef] [PubMed]
- Di Noto, R.; Lo Pardo, C.; Schiavone, E.M.; Ferrara, F.; Manzo, C.; Vacca, C.; Del Vecchio, L. All-trans retinoic acid (ATRA) and the regulation of adhesion molecules in acute myeloid leukemia. Leuk. Lymphoma Inf. Healthc. 1996, 21, 201–209. Available online: https://pubmed.ncbi.nlm.nih.gov/8726400/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- De Santis, G.C.; Tamarozzi, M.D.B.; Sousa, R.B.; Moreno, S.E.; Secco, D.; Garcia, A.B.; Lima, A.S.G.; Faccioli, L.H.; Falcão, R.P.; Cunha, F.Q.; et al. Adhesion molecules and differentiation syndrome: Phenotypic and functional analysis of the effect of ATRA, As2O3, phenylbutyrate, and G-CSF in acute promyelocytic leukemia. Haematologica 2007, 92, 1615–1622. Available online: https://pubmed.ncbi.nlm.nih.gov/18055984/ (accessed on 18 May 2022). [CrossRef]
- Najmaddini, H.; Hassanshahi, G.; Ostadebrahimi, H.; Barkhordari, H.; Mashayekhi, H.; Nazari, M.; Moogooei, M.; Arababadi, Y.S.; Peighambari, F.; Karimabad, M.N. Overproduction of CXC chemokines CXCL1, CXCL9, CXCL10 and CXCL12 in β-thalassemia major or patients. Ann. Saudi Med. 2014, 34, 122–127. Available online: https://pubmed.ncbi.nlm.nih.gov/24894780/ (accessed on 18 May 2022). [CrossRef]
- Luesink, M.; Pennings, J.L.A.; Wissink, W.M.; Linssen, P.C.M.; Muus, P.; Pfundt, R.; de Witte, T.J.M.; van der Reijden, B.A.; Jansen, J.H. Chemokine induction by all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia: Triggering the differentiation syndrome. Blood 2009, 114, 5512–5521. Available online: https://pubmed.ncbi.nlm.nih.gov/19828696/ (accessed on 18 May 2022). [CrossRef]
- Albanesi, J.; Noguera, N.; Banella, C.; Colangelo, T.; De Marinis, E.; Leone, S.; Palumbo, O.; Voso, M.; Ascenzi, P.; Nervi, C.; et al. Transcriptional and Metabolic Dissection of ATRA-Induced Granulocytic Differentiation in NB4 Acute Promyelocytic Leukemia Cells. Cells 2020, 9, 2423. Available online: https://www.mdpi.com/2073-4409/9/11/2423/htm (accessed on 18 May 2022).
- Mueller, B.U.; Pabst, T.; Fos, J.; Petkovic, V.; Fey, M.F.; Asou, N.; Buergi, U.; Tenen, D.G. ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU.1 expression. Blood 2006, 107, 3330–3338. Available online: https://pubmed.ncbi.nlm.nih.gov/16352814/ (accessed on 18 May 2022). [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Caprari, P.; Massimi, S.; Sorrentino, F.; Maffei, L.; Riganò, R. Phenotypical and functional abnormalities of circulating neutrophils in patients with β-thalassemia. Ann. Hematol. 2020, 99, 2265–2277. Available online: https://pubmed.ncbi.nlm.nih.gov/32803313/ (accessed on 18 May 2022). [CrossRef]
- Siwaponanan, P.; Siegers, J.Y.; Ghazali, R.; Ng, T.; Mccoll, B.; Ng, G.Z.W.; Sutton, P.; Wang, N.; Ooi, I.; Thiengtavor, C.; et al. Reduced PU.1 expression underlies aberrant neutrophil maturation and function in b-Thalassemia mice and patients. Blood 2017, 129, 3087–3099. Available online: https://pubmed.ncbi.nlm.nih.gov/28325862/ (accessed on 18 May 2022). [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Malignancy | Reference | Patients (n) | Sex/Age | Hemoglobinopathy | Splenectomy | Clinical Presentation | Therapy/Outcome |
|---|---|---|---|---|---|---|---|
| HL | Balestrazzi and Butturini,1985 [5] | 1 | M/10 | BTM | ND | Lymphocyte rich cHL (CS II2B) | 6MOPP + RT DF at 3 years |
| Thapa et al., 2009 [6] | 1 | F/7 | BTM | No | Mixed cellularity cHL; stage IV | 6 COPP, Complete remission | |
| Jabr et al., 2006 [7] | 1 | M/28 | BTI | No | Mixed cellularity cHL; IIIa | ABVD, complete remission | |
| Ahad-Aziz Qureshi et al., 2019 [8] | 1 | M/21 | BTM | Yes | Lymphocyte-rich cHL | Not reported | |
| NHL | Tozzi-Cecchetti, 1979 et al. [9] | 1 | M/11 | BTM | No | Diffuse follicular center lymphoma | 1 VMP Death at 3 months |
| Schilirò et al., 1979 [10] | 1 | M/4 | BTM | No | NHL with bone marrow and CNS involvement. | 1 HOP + 2 IT-MTX + prophylactic CI. Death after relapse. | |
| Otrock et al., 2006 [11] | 1 | F/25 | BTM | No | DLBCL | 4 CNOP + RT. Complete remission, 6 years’ follow-up | |
| Ricchi et al., 2004 [12] | 1 | F/35 | BTM, HCV+ | Yes | Systemic High-grade NHL with cardiac localization | 3 RTX + surgery. Death for hemopericardium with cardiac tamponade | |
| Triantos et al., 2013 [13] | 1 | ND | BTM | ND | NHL | Not reported | |
| Chehal et al., 2002 [14] | 1 | F/62 | BTI | No | Large B cell lymphoma with bone marrow involvement | 6 CVP + 4 RTX. Complete remission | |
| ALL | Tuğcu et al., 2014 [15] | 1 | M/12 | BTI | No | B-ALL | Chemotherapy Complete remission |
| Kottaridis et al., 2000 [16] | 1 | M/34 | BTM | No | T-ALL | Chemotherapy + alloHSCT Complete remission at 1 year | |
| Goyal et al., 2018 [17] | 1 | M/22 | BTM | No | ALL in the context of HIV and HTLV-I infection | Death before treatment | |
| Sherief et al., 2015 [18] | 1 | M/11 | BTM, HCV+ | No | B-ALL | Chemotherapy Complete remission | |
| Palomo-Colli et al., 2018 [19] | 1 | M/6 | BTM | Yes | ALL-L1, B phenotype | Chemotherapy CNS radiotherapy 7y in Complete remission | |
| AML | Felici et al., 1988 [20] | 1 | M/6 | BTM | No | AML-M4 | Chemotherapy Death 10d after induction |
| Romani et al., 2007 [21] | 1 | M/32 | BTM, HCV+ | No | APL | ATRA+ idarubicine (induction) ATRA + idarubicine + ara-c (consolidation) Death for liver failure | |
| Histiocytic disorder | De Soccio et al., 2020 [22] | 1 | F/4 | BTM | No | Solitary xantogranuloma of the hypopharynx and cutaneous mastocytosis | Surgery. Complete remission at 11 months |
| MM | Arora & Ganapathi, 2020 [23] | 1 | M/46 | BTI | ND | IgG lambda myeloma | Not reported |
| MPN | Voskaridou et al., 2002 [24] | 1 | M/32 | BTI | Yes | BCR/ABL+ CML with marked thrombocytosis | Hydroxyurea + Anagrelide |
| Wu et al., 2020 [25] | 1 | F/24 | BTI | Yes | ET | Observation | |
| Various | Alavi et al., 2013 [26] | 2 | F/16 | BTM, HCV+ | No | HL nodular sclerosis, IIA | 6 alternating MOPP/ABVD + RT; CR at 10y |
| F/13 | BTI | No | CML, progressed to AML | Imatinib, CT. Death at 3 months | |||
| Benetatos et al., 2008 [27] | 2 | M/37 | BTI, HCV+ | Yes | Marginal zone lymphoma, IIIa | Watch and wait. Stable disease at 2y | |
| F/52 | BTI | No | Nodular lymphocytic predominant HL IIIa | ABVD complete remission | |||
| Karimi et al., 2009 [28] | 10 | M/10 | BTM | Yes | cHL | Chemotherapy | |
| M/6 | BTM | No | cHL | Chemotherapy | |||
| M/20 | BTM, HCV+ | No | CML | Chemotherapy | |||
| M/16 | BTM | Yes | ALL-preB | Chemotherapy | |||
| M/6 | BTI | Yes | AML-M4 | Chemotherapy | |||
| M/12 | BTI | No | ALL-T | Chemotherapy | |||
| M/23 | BTM, HCV+ | Yes | DLBCL | Chemotherapy | |||
| F/28 | BTM, HCV+ | Yes | ALL-T | Chemotherapy | |||
| M/25 | BTM, HCV+ | Yes | NHL | Chemotherapy | |||
| F/24 | BTM | Yes | cHL | Chemotherapy | |||
| Zurlo et al., 1989 [29] | 5 | ND | BTM | ND | 2 NHL; 2 ALL; 1 AML | Not reported |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrino, C.; Dragonetti, G.; Chiusolo, P.; Rossi, M.; Orlando, N.; Teofili, L. Acute Promyelocytic Leukemia in a Woman with Thalassemia Intermedia: Case Report and Review of Literature on Hematological Malignancies in β-Thalassemia Patients. Hematol. Rep. 2022, 14, 310-321. https://doi.org/10.3390/hematolrep14040045
Pellegrino C, Dragonetti G, Chiusolo P, Rossi M, Orlando N, Teofili L. Acute Promyelocytic Leukemia in a Woman with Thalassemia Intermedia: Case Report and Review of Literature on Hematological Malignancies in β-Thalassemia Patients. Hematology Reports. 2022; 14(4):310-321. https://doi.org/10.3390/hematolrep14040045
Chicago/Turabian StylePellegrino, Claudio, Giulia Dragonetti, Patrizia Chiusolo, Monica Rossi, Nicoletta Orlando, and Luciana Teofili. 2022. "Acute Promyelocytic Leukemia in a Woman with Thalassemia Intermedia: Case Report and Review of Literature on Hematological Malignancies in β-Thalassemia Patients" Hematology Reports 14, no. 4: 310-321. https://doi.org/10.3390/hematolrep14040045
APA StylePellegrino, C., Dragonetti, G., Chiusolo, P., Rossi, M., Orlando, N., & Teofili, L. (2022). Acute Promyelocytic Leukemia in a Woman with Thalassemia Intermedia: Case Report and Review of Literature on Hematological Malignancies in β-Thalassemia Patients. Hematology Reports, 14(4), 310-321. https://doi.org/10.3390/hematolrep14040045