
The Co-Occurrence of Autism Spectrum Disorder and Aarskog–Scott Syndrome in an Accomplished Young Man

 , , , and
, , , and
Abstract
1. Introduction
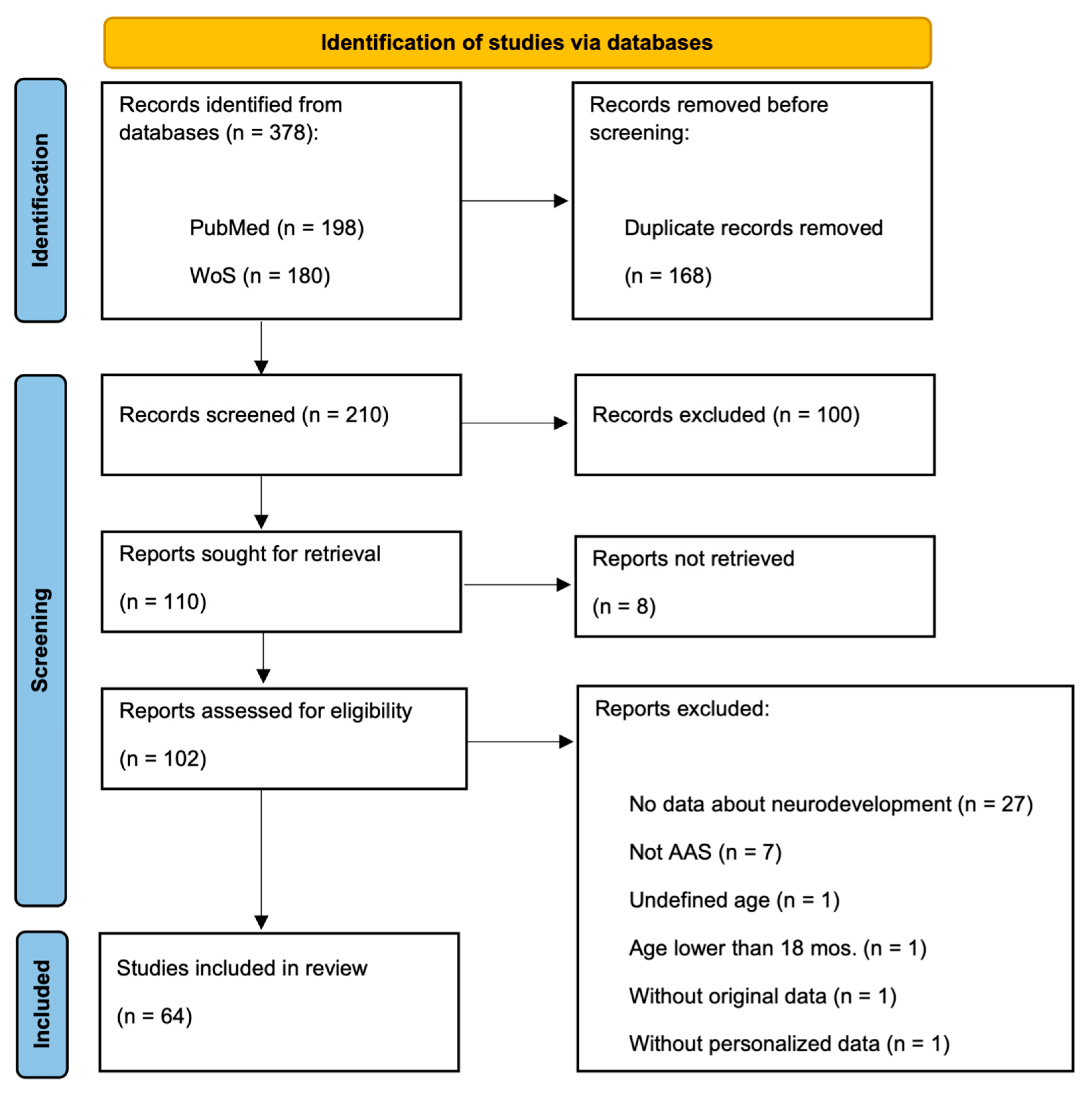
2. Materials and Methods
2.1. Genetic Testing
2.2. Clinical and Behavioral Assessment
3. Results
3.1. Case Presentation
3.2. Molecular Findings
3.3. Clinical and Behavioral Assessment
3.3.1. ASD Diagnostics
3.3.2. Cognitive Development
3.3.3. Adaptive Functioning
3.4. Assessment of Comorbid Disorders
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
| ADI-R Domain | Score | Threshold |
|---|---|---|
| Qualitative impairments in reciprocal social interaction | 25 | 10 |
| Qualitative impairments in communication (verbal) | 9 | 8 |
| Repetitive behaviors and stereotyped patterns | 10 | 3 |
| ADOS-2 Scale | Item | Score |
|---|---|---|
| Social affect (SA) | ||
| Language and communication | ||
| Reporting of events | A-7 | 0 |
| Conversation | A-8 | 0 |
| Descriptive, conventional, or informational gestures | A-9 | 1 |
| Reciprocal social interaction | ||
| Unusual eye contact | B-1 | 2 |
| Facial expressions directed to others | B-2 | 1 |
| Shared enjoyment in interaction | B-4 | 1 |
| Quality of social overtures | B-7 | 1 |
| Quality of social response | B-9 | 1 |
| Amount of reciprocal social communication | B-10 | 0 |
| Overall quality of rapport | B-11 | 0 |
| SA total: | 7 | |
| Restricted and repetitive behavior (RRB) | ||
| Play, stereotyped behaviors, and restricted interests | ||
| Stereotyped/idiosyncratic use of words or phrases | A-4 | 1 |
| Unusual sensory interest in play material/person | D-1 | 0 |
| Hand and finger and other complex mannerisms | D-2 | 0 |
| Excessive interest in or references to unusual or highly specific topics or objects or repetitive behaviors | D-4 | 1 |
| RRB total: | 2 | |
| Overall total: | 9 | |
| Leiter-3 Subtest | Scaled Score | Descriptive Classification | ||
| Figure Ground | 9 | Average | ||
| Form Completion | 12 | Average | ||
| Classification and Analogies | 14 | Above Average | ||
| Leiter-3 Batteries | Scaled Score | 95% CI | Percentile Rank | Descriptive Classification |
| Nonverbal IQ | 115 | [109, 121] | 84 | Above Average |
| Nonverbal Memory | 107 | [97, 117] | 68 | Average |
| Processing Speed | 92 | [85, 99] | 30 | Average |
| UNIT-2 Subtest | Scaled Score | Descriptive Classification | ||
| Symbolic Memory | 9 | Average | ||
| Nonsymbolic Quantity | 12 | Average | ||
| Analogic Reasoning | 11 | Average | ||
| Spatial Memory | 14 | Above Average | ||
| Numeral Series | 10 | Average | ||
| Cube Design | 15 | Superior | ||
| UNIT-2 Composite | Index Scaled Score | 95% CI | Percentile Rank | Descriptive Classification |
| Memory | 109 | [101, 116] | 73 | Average |
| Reasoning | 117 | [111, 122] | 87 | Above Average |
| Quantitative | 106 | [101, 111] | 66 | Average |
| Full Scale Battery | 113 | [109, 117] | 81 | Above Average |
| Vineland-II Domain | Standard Score | Percentile Rank | Descriptive Classification |
| Communication | 87 | 19 | Adequate |
| Daily living skills | 58 | <1 | Low, mild deficit |
| Socialization | 58 | <1 | Low, mild deficit |
| Adaptive behavior composite | 65 | 1 | Low, mild deficit |
| Vineland-II Maladaptive Domain | V-Score | Descriptive Classification | |
| Internalizing | 22 | Clinically significant | |
| Externalizing | 19 | Elevated | |
| Maladaptive behavior index | 21 | Clinically significant | |
| Subscale | T-Score | Percentile | Range |
|---|---|---|---|
| Syndrome Scale | |||
| Anxious/Depressed | 60 | 84 | Normal |
| Withdrawn/Depressed | 60 | 84 | Normal |
| Somatic Complaints | 61 | 86 | Normal |
| Social Problems | 77 | >98 | Clinical |
| Thought Problems | 66 | 94 | Subclinical |
| Attention Problems | 64 | 92 | Normal |
| Rule-Breaking Behavior | 57 | 75 | Normal |
| Aggressive Behavior | 58 | 78 | Normal |
| Internalizing Behavior | 62 | - | Subclinical |
| Externalizing Behavior | 58 | - | Normal |
| Total | 64 | - | Clinical |
| Competence Scale | |||
| Activities | 45 | 31 | Normal |
| Social | 50 | 50 | Normal |
| School | 34 | 6 | Subclinical |
| Total | 43 | - | Normal |
| DSM-Oriented Scale | |||
| Affective Problems | 61 | 86 | Normal |
| Anxiety Problems | 67 | 94 | Subclinical |
| Somatic Problems | 61 | 84 | Normal |
| Attention Deficit/Hyperactivity Problems | 67 | 95 | Subclinical |
| Oppositional Defiant Problems | 58 | 78 | Normal |
| Conduct Problems | 56 | 72 | Normal |
| Index/Scale | T-Score | 90% CI | Percentile | Descriptive Classification |
|---|---|---|---|---|
| Inhibit | 64 | [58; 70] | 92 | Mildly elevated |
| Self-Monitor | 63 | [56; 70] | 93 | Mildly elevated |
| Behavior Regulation Index | 65 | [60; 70] | 91 | Mildly elevated |
| Shift | 72 | [64; 78] | 96 | Potentially clinically elevated |
| Emotional Control | 72 | [67; 75] | 96 | Clinically elevated |
| Emotion Regulation Index | 73 | [69; 77] | 96 | Clinically elevated |
| Initiate | 59 | [53; 65] | 85 | Not elevated |
| Working Memory | 65 | [60; 70] | 92 | Potentially clinically elevated |
| Plan/Organize | 67 | [62; 72] | 90 | Potentially clinically elevated |
| Task-Monitor | 62 | [57; 67] | 90 | Mildly elevated |
| Organization of Materials | 60 | [55; 65] | 86 | Mildly elevated |
| Cognitive Regulation Index | 65 | [62; 68] | 90 | Potentially clinically elevated |
| Global Executive Composite | 69 | [67; 71] | 95 | Potentially clinically elevated |
| Index/Scale | T-Score | 90% CI | Percentile | Descriptive Classification |
|---|---|---|---|---|
| Inhibit | 67 | [61; 73] | 94 | Potentially clinically elevated |
| Self-Monitor | 74 | [81; 67] | 99 | Clinically elevated |
| Behavior Regulation Index | 70 | [65; 75] | 96 | Clinically elevated |
| Shift | 76 | [70; 82] | 98 | Clinically elevated |
| Emotional Control | 84 | [79; 89] | >99 | Clinically elevated |
| Emotion Regulation Index | 83 | [79; 87] | >99 | Clinically elevated |
| Initiate | 55 | [49; 61] | 71 | Not elevated |
| Working Memory | 67 | [62; 72] | 94 | Potentially clinically elevated |
| Plan/Organize | 69 | [64; 74] | 92 | Potentially clinically elevated |
| Task-Monitor | 73 | [68; 78] | >99 | Clinically elevated |
| Organization of Materials | 52 | [47; 57] | 65 | Not elevated |
| Cognitive Regulation Index | 65 | [62; 68] | 90 | Potentially clinically elevated |
| Global Executive Composite | 76 | [74; 78] | 99 | Clinically elevated |
| Index/Scale | T-Score | 90% CI | Percentile | Descriptive Classification |
|---|---|---|---|---|
| Inhibit | 62 | [56; 68] | 85 | Mildly elevated |
| Self-Monitor | 56 | [49; 63] | 79 | Not elevated |
| Behavior Regulation Index | 61 | [56; 66] | 83 | Mildly elevated |
| Shift | 86 | [92; 80] | >99 | Clinically elevated |
| Emotional Control | 78 | [71; 85] | 99 | Clinically elevated |
| Emotion Regulation Index | 85 | [80; 90] | >99 | Clinically elevated |
| Task Compilation | 61 | [55; 67] | 84 | Mildly elevated |
| Working Memory | 69 | [63; 75] | 96 | Potentially clinically elevated |
| Plan/Organize | 62 | [57; 67] | 88 | Mildly elevated |
| Cognitive Regulation Index | 65 | [62; 68] | 90 | Potentially clinically elevated |
| Global Executive Composite | 70 | [67; 73] | 96 | Clinically elevated |
| Conners 3 Content Scale | T-Score | Range | |
| Inattention | 59 | Average Score | |
| Hyperactivity/Impulsivity | 58 | Average Score | |
| Learning Problems | 71 | Very Elevated Score | |
| Executive Functioning | 47 | Average Score | |
| Aggression | 48 | Average Score | |
| Peer Relations | 73 | Very Elevated Score | |
| Conners-3 Global Index Total | 59 | Average Score | |
| DSM-5 Symptom Scale | T-Score | Range | Symptom Count Requirements |
| ADHD Inattention | 56 | Average Score | 1 from 6 |
| ADHD Hyperactivity/Impulsivity | 43 | Average Score | 2 from 6 |
| Conduct Disorder | 57 | Average Score | 0 from 3 |
| Oppositional Defiant Disorder | 44 | Average Score | 1 from 4 |
Appendix B
References
- Orrico, A.; Galli, L.; Clayton-Smith, J.; Fryns, J.P. Clinical utility gene card for: Aarskog–Scott syndrome (faciogenital dysplasia)–update 2015. Eur. J. Pediatr. 2015, 23, 558. [Google Scholar] [CrossRef]
- Li, S.; Tian, A.; Wen, Y.; Gu, W.; Li, W.; Qiao, X.; Zhang, C.; Luo, X. FGD1-related Aarskog–Scott syndrome: Identification of four novel variations and a literature review of clinical and molecular aspects. Eur. J. Pediatr. 2024, 183, 2257–2272. [Google Scholar] [CrossRef]
- Teebi, A.S.; Rucquoi, J.K.; Meyn, M.S. Aarskog syndrome: Report of a family with review and discussion of nosology. Am. J. Med. Genet. 1993, 46, 501–509. [Google Scholar] [CrossRef]
- Drumond, V.Z.; Salgado, L.S.; Salgado, C.S.; de Lima Oliveira, V.A.; de Assis, E.M.; Ribeiro, M.C.; Valadao, A.F.; Orrico, A. The prevalence of clinical features in patients with Aarskog–Scott syndrome and assessment of genotype-phenotype correlation: A systematic review. Genet. Res. 2021, 2021, 6652957. [Google Scholar]
- Zhu, Y.; Chen, Q.; Lin, H.; Lu, H.; Qu, Y.; Yan, Q.; Wang, C. FGD1 Variant Associated with Aarskog–Scott Syndrome. Front. Pediatr. 2022, 10, 888923. [Google Scholar] [CrossRef]
- Olson, M.F.; Pasteris, N.G.; Gorski, J.L.; Hall, A. Faciogenital dysplasia protein (FGD1) and Vav, two related proteins required for normal embryonic development, are upstream regulators of Rho GTPases. Curr. Biol. 1996, 6, 1628–1633. [Google Scholar] [CrossRef]
- Jabalameli, M.R.; Briceno, I.; Martinez, J.; Pengelly, R.J.; Ennis, S.; Collins, A. Aarskog-Scott syndrome: Phenotypic and genetic heterogeneity. AIMS Genet. 2016, 3, 49–59. [Google Scholar] [CrossRef]
- Orrico, A.; Galli, L.; Faivre, L.; Clayton-Smith, J.; Azzarello-Burri, S.M.; Hertz, J.M.; Jacquemont, S.; Taurisano, R.; Carrera, I.A.; Tarantino, E.; et al. Aarskog-Scott Syndrome: Clinical Update and Report of Nine Novel Mutations of the FGD1 Gene. Am. J. Med. Genet. 2010, A152a, 313–318. [Google Scholar] [CrossRef]
- Assumpcao, F.; Santos, R.C.; Rosario, M.; Mercadante, M. Brief report: Autism and Aarskog syndrome. J. Autism Dev. Disord. 1999, 29, 179–181. [Google Scholar] [CrossRef]
- De Wolf, V.; Crepel, A.; Schuit, F.; van Lommel, L.; Ceulemans, B.; Steyaert, J.; Seuntjens, E.; Peeters, H.; Devriendt, K. A complex Xp11.22 deletion in a patient with syndromic autism: Exploration of FAM120C as a positional candidate gene for autism. Am. J. Med. Genet. Part A 2014, 164A, 3035–3041. [Google Scholar] [CrossRef]
- Orrico, A.; Galli, L.; Buoni, S.; Hayek, G.; Luchetti, A.; Lorenzini, S.; Zappella, M.; Pomponi, M.G.; Sorrentino, V. Attention-deficit/hyperactivity disorder (ADHD) and variable clinical expression of Aarskog-Scott syndrome due to a novel FGD1 gene mutation (R408Q). Am. J. Med. Genet. A 2005, 135, 99–102. [Google Scholar] [CrossRef]
- Sugarman, G.I.; Rimoin, D.L.; Lachman, R.S. The facial-digital-genital (Aarskog) syndrome. Am. J. Dis. Child. 1973, 126, 248–252. [Google Scholar] [CrossRef]
- Logie, L.J.; Porteous, M.E. Intelligence and development in Aarskog syndrome. Arch. Dis. Child. 1998, 79, 359–360. [Google Scholar] [CrossRef]
- Bottani, A.; Orrico, A.; Galli, L.; Karam, O.; Haenggeli, C.A.; Ferey, S.; Conrad, B. Unilateral focal polymicrogyria in a patient with classical Aarskog-Scott syndrome due to a novel missense mutation in an evolutionary conserved RhoGEF domain of the faciogenital dysplasia gene FGD1. Am. J. Med. Genet. A 2007, 143A, 2334–2338. [Google Scholar] [CrossRef]
- Liang, Y.; Wu, H.; He, X.; He, X. Case Report: Aarskog-scott syndrome caused by FGD1 gene variation: A family study. Front. Genet. 2022, 13, 932073. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rutter, M.; Le Couteur, A.; Lord, C. Autism Diagnostic Interview-Revised; Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Lord, C.; Rutter, M.; DiLavore, P.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule–2nd Edition (ADOS-2); Western Psychological Services: Los Angeles, CA, USA, 2012. [Google Scholar]
- Bracken, B.; McCallum, R.S. Universal Nonverbal Intelligence Test, 2nd ed.; PRO-ED: Austin, TX, USA, 2016. [Google Scholar]
- Roid, G.H.; Miller, L.J.; Pomplun, M.; Koch, C. Leiter International Performance Scale, 3rd ed.; Stoelting Company: Wood Dale, IL, USA, 2013. [Google Scholar]
- Sparrow, S.S.; Cicchetti, D.V.; Balla, D.A. Vineland Adaptive Behavior Scales, 2nd ed.; American Guidance Service: Circle Pines, MN, USA, 2005. [Google Scholar]
- Lai, M.C.; Kassee, C.; Besney, R.; Bonato, S.; Hull, L.; Mandy, W.; Szatmari, P.; Ameis, S.H. Prevalence of co-occurring mental health diagnoses in the autism population: A systematic review and meta-analysis. Lancet Psychiatry 2019, 6, 819–829. [Google Scholar] [CrossRef]
- Achenbach, T.M.; Rescorla, L.A. Manual for the ASEBA School-Age Forms & Profiles; University of Vermont, Research Center for Children, Youth, and Families: Burlington, VT, USA, 2001. [Google Scholar]
- Gioia, G.A.; Isquith, P.K.; Guy, S.C.; Kenworthy, L. Behavior Rating Inventory of Executive Function®, 2nd ed.; PAR Inc.: Lutz, FL, USA, 2015. [Google Scholar]
- Conners, C.K. Conners Third Edition (Conners 3); Western Psychological Services: Los Angeles, CA, USA, 2008. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; Text Rev.; American Psychiatric Association Publishing: Washington, DC, USA, 2022. [Google Scholar]
- Altıncık, A.; Kaname, T.; Demir, K.; Böber, E. A novel mutation in a mother and a son with Aarskog-Scott syndrome. J. Pediatr. Endocrinol. Metab. 2013, 26, 385–388. [Google Scholar] [CrossRef]
- Talantseva, O.I.; Portnova, G.V.; Romanova, R.S.; Martynova, D.A.; Sysoeva, O.V.; Grigorenko, E.L. Does the Potocki–Lupski Syndrome Convey the Autism Spectrum Disorder Phenotype? Case Report and Scoping Review. J. Pers. Med. 2023, 13, 439. [Google Scholar] [CrossRef]
- Praticò, A.D.; Falsaperla, R.; Rizzo, R.; Ruggieri, M.; Verrotti, A.; Pavone, P. A New Patient with Potocki-Lupski Syndrome: A Literature Review. J. Pediatr. Genet. 2018, 7, 29–34. [Google Scholar]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef]
- Melnick, M.; Shields, E.D. Aarskog syndrome: New oral-facial findings. Clin. Genet. 1976, 9, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.B.; Stanton, A. Aarskog syndrome: A case report and literature review. Optometry 2008, 79, 371–377. [Google Scholar] [CrossRef]
- Talantseva, O.I.; Romanova, R.S.; Shurdova, E.M.; Dolgorukova, T.A.; Sologub, P.S.; Titova, O.S.; Kleeva, D.F.; Grigorenko, E.L. The global prevalence of autism spectrum disorder: A three-level meta-analysis. Front. Psychiatry 2023, 14, 1071181. [Google Scholar] [CrossRef]
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Orrico, A.; Galli, L.; Cavaliere, M.L.; Garavelli, L.; Fryns, J.P.; Crushell, E.; Rinaldi, M.M.; Medeira, A.; Sorrentino, V. Phenotypic and molecular characterisation of the Aarskog-Scott syndrome: A survey of the clinical variability in light of FGD1 mutation analysis in 46 patients. Eur. J. Hum. Genet. 2004, 12, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.; Egger, J.I.; Hoogeboom, A.J. X-linked Aarskog syndrome: Report on a novel FGD1 gene mutation. Executive dysfunction as part of the behavioural phenotype. Genet. Couns. 2012, 23, 157–167. [Google Scholar]
- Diluna, M.L.; Amankulor, N.M.; Johnson, M.H.; Gunel, M. Cerebrovascular disease associated with Aarskog-Scott syndrome. Neuroradiology 2007, 49, 457–461. [Google Scholar] [CrossRef]
- Salari, N.; Ghasemi, H.; Abdoli, N.; Rahmani, A.; Shiri, M.H.; Hashemian, A.H.; Akbari, H.; Mohammadi, M. The global prevalence of ADHD in children and adolescents: A systematic review and meta-analysis. Ital. J. Pediatr. 2023, 49, 48. [Google Scholar] [CrossRef]
- Song, P.; Zha, M.; Yang, Q.; Zhang, Y.; Li, X.; Rudan, I. The prevalence of adult attention-deficit hyperactivity disorder: A global systematic review and meta-analysis. J. Glob. Health 2021, 11, 04009. [Google Scholar] [CrossRef]
- Kaname, T.; Yanagi, K.; Okamoto, N.; Naritomi, K. Neurobehavioral disorders in patients with Aarskog-Scott syndrome affected by novel FGD1 mutations. Am. J. Med. Genet. A 2006, 140, 1331–1332. [Google Scholar] [CrossRef]
- Fryns, J.P. Aarskog syndrome: The changing phenotype with age. Am. J. Med. Genet. 1992, 43, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Craig, F.; Margari, F.; Legrottaglie, A.R.; Palumbi, R. A review of executive function deficits in autism spectrum disorder and attention-deficit/hyperactivity disorder. Neuropsychiatr. Dis. Treat. 2016, 12, 1191–1202. [Google Scholar]
- Arnold, L.E.; Hodgkins, P.; Kahle, J.; Madhoo, M.; Kewley, G. Long-Term Outcomes of ADHD: Academic Achievement and Performance. J. Atten. Disord. 2020, 24, 73–85. [Google Scholar] [CrossRef]
- Gabbay-Dizdar, N.; Ilan, M.; Meiri, G.; Faroy, M.; Michaelovski, A.; Flusser, H.; Menashe, I.; Koller, J.; Zachor, D.A.; Dinstein, I. Early diagnosis of autism in the community is associated with marked improvement in social symptoms within 1–2 years. Autism 2022, 26, 1353–1363. [Google Scholar] [CrossRef]
- Al-Semari, A.; Wakil, S.M.; Al-Muhaizea, M.A.; Dababo, M.; Al-Amr, R.; Alkuraya, F.; Meyer, B.F. Novel FGD1 mutation underlying Aarskog-Scott syndrome with myopathy and distal arthropathy. Clin. Dysmorphol. 2013, 22, 13–17. [Google Scholar] [CrossRef]
- Andrassy, R.J.; Murthy, S.; Woolley, M.M. Aarskog syndrome: Significance for the surgeon. J. Pediatr. Surg. 1979, 14, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Bae, G.Y.; Kim, M.S.; Kim, J.Y.; Jang, J.H.; Lee, S.M.; Cho, S.Y.; Jin, D.K. The First Korean Family with Aarskog-Scott Syndrome Harboring a Novel Mutation in FGD1 Diagnosed via Targeted Gene Panel Sequencing. Ann. Clin. Lab. Sci. 2020, 50, 691–698. [Google Scholar] [PubMed]
- Bawle, E.; Tyrkus, M.; Lipman, S.; Bozimowski, D. Aarskog syndrome: Full male and female expression associated with an X-autosome translocation. Am. J. Med. Genet. 1984, 17, 595–602. [Google Scholar] [CrossRef]
- Bayat, A.; Krett, B.; Dunø, M.; Torring, P.M.; Vissing, J. Novel truncating variants in FGD1 detected in two Danish families with Aarskog-Scott syndrome and myopathic features. Am. J. Med. Genet. A 2022, 188, 2251–2257. [Google Scholar] [CrossRef]
- Bedoyan, J.K.; Friez, M.J.; DuPont, B.; Ahmad, A. First case of deletion of the faciogenital dysplasia 1 (FGD1) gene in a patient with Aarskog-Scott syndrome. Eur. J. Med. Genet. 2009, 52, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Berman, P.; Desjardins, C.; Fraser, F.C. The inheritance of the Aarskog facial-digital-genital syndrome. J. Pediatr. 1975, 86, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.; Cree, J.; Mann, T. Aarskog’s syndrome. Arch. Dis. Child. 1980, 55, 706–710. [Google Scholar] [CrossRef]
- Braiotta, F.; Paglia, M.; Mummolo, S. Aarskog-scott syndrome (AAS): A case report. Eur. J. Paediatr. Dent. 2023, 24, 238–240. [Google Scholar]
- Brodsky, M.C.; Keppen, L.D.; Rice, C.D.; Ranells, J.D. Ocular and systemic findings in the Aarskog (facial-digital-genital) syndrome. Am. J. Ophthalmol. 1990, 109, 450–456. [Google Scholar] [CrossRef]
- Escobar, V.; Weaver, D.D. Aarskog syndrome. New findings and genetic analysis. JAMA 1978, 240, 2638–2641. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.; Tsukahara, M.; Mito, H.; Yoshii, H.; Uchida, M.; Matsuo, K.; Kajii, T. Congenital heart defects in Aarskog syndrome. Am. J. Med. Genet. 1994, 50, 318–322. [Google Scholar] [CrossRef]
- Fryns, J.P.; Macken, J.; Vinken, L.; Igodt-Ameye, L.; van den Berghe, H. The Aarskog syndrome. Hum. Genet. 1978, 42, 129–135. [Google Scholar] [CrossRef]
- Funderburk, S.J.; Crandall, B.F. The Aarskog syndrome in three brothers. Clin. Genet. 1974, 6, 119–124. [Google Scholar] [CrossRef]
- Furukawa, C.T.; Hall, B.D.; Smith, D.W. The Aarskog syndrome. J. Pediatr. 1972, 81, 1117–1122. [Google Scholar] [CrossRef]
- Gahlot, D.; Aggarwal, M. Anaesthetic considerations in Aarskog Scott Syndrome: A syndrome new to our understanding. Saudi J. Anaesth. 2021, 15, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Grier, R.E.; Farrington, F.H.; Kendig, R.; Mamunes, P. Autosomal dominant inheritance of the Aarskog syndrome. Am. J. Med. Genet. 1983, 15, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Guion-Almeida, M.L.; Richieri-Costa, A. Aarskog syndrome in a Brazilian boy born to consanguineous parents. Am. J. Med. Genet. 1992, 43, 808–810. [Google Scholar] [CrossRef]
- Hamzeh, A.R.; Saif, F.; Nair, P.; Binjab, A.J.; Mohamed, M.; Al-Ali, M.T.; Bastaki, F. A novel, putatively null, FGD1 variant leading to Aarskog-Scott syndrome in a family from UAE. BMC Pediatr. 2017, 17, 31. [Google Scholar] [CrossRef]
- Hoo, J.J. The Aarskog (facio-digito-genital) syndrome. Clin. Genet. 1979, 16, 269–276. [Google Scholar] [CrossRef]
- Kessel, I.; German, A.; Peleg, A.; Regeneron Genetics Center; Gonzaga-Jauregui, C.; Paperna, T.; Ekhilevitch, N.; Kurolap, A.; Baris Feldman, H.; Sagi-Dain, L. A novel truncating variant in the FGD1 gene associated with Aarskog-Scott syndrome in a family previously diagnosed with Tel Hashomer camptodactyly. Am. J. Med. Genet. A 2021, 185, 3161–3166. [Google Scholar] [CrossRef]
- Kodama, M.; Fujimoto, S.; Namikawa, T.; Matsuda, I. Aarskog syndrome with isolated growth hormone deficiency. Eur. J. Pediatr. 1981, 135, 273–276. [Google Scholar] [CrossRef]
- Menezes, A.H.; Traynelis, V.C. Pediatric cervical kyphosis in the MRI era (1984–2008) with long-term follow up: Literature review. Childs Nerv. Syst. 2022, 38, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Meschede, D.; Rolf, C.; Neugebauer, D.C.; Horst, J.; Nieschlag, E. Sperm acrosome defects in a patient with Aarskog-Scott syndrome. Am. J. Med. Genet. 1996, 66, 340–342. [Google Scholar] [CrossRef]
- Mikelsaar, R.V.; Lurie, I.W. Atypical case of Aarskog syndrome. J. Med. Genet. 1992, 29, 349–350. [Google Scholar] [CrossRef]
- Nayak, R.B.; Ambika, L.; Bhogale, G.S.; Pandurangi, A. Mania with Aarskog-Scott syndrome. Indian Pediatr. 2012, 49, 327–328. [Google Scholar] [PubMed]
- Nielsen, K.B. Aarskog syndrome in a Danish family: An illustration of the need for dysmorphology in paediatrics. Clin. Genet. 1988, 33, 315–317. [Google Scholar] [CrossRef]
- Oberiter, V.; Lovrencić, M.K.; Schmutzer, L.; Kraus, O. The Aarskog syndrome. Acta Paediatr. Scand. 1980, 69, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Orrico, A.; Galli, L.; Falciani, M.; Bracci, M.; Cavaliere, M.L.; Rinaldi, M.M.; Musacchio, A.; Sorrentino, V. A mutation in the pleckstrin homology (PH) domain of the FGD1 gene in an Italian family with faciogenital dysplasia (Aarskog-Scott syndrome). FEBS Lett. 2000, 478, 216–220. [Google Scholar] [CrossRef]
- Orrico, A.; Galli, L.; Obregon, M.G.; de Castro Perez, M.F.; Falciani, M.; Sorrentino, V. Unusually severe expression of craniofacial features in Aarskog-Scott syndrome due to a novel truncating mutation of the FGD1 gene. Am. J. Med. Genet. A 2007, 143A, 58–63. [Google Scholar] [CrossRef]
- Parıltay, E.; Hazan, F.; Ataman, E.; Demir, K.; Etlik, Ö.; Özbek, E.; Özkan, B. A novel splice site mutation of FGD1 gene in an Aarskog-Scott syndrome patient with a large anterior fontanel. J. Pediatr. Endocrinol. Metab. 2016, 29, 1111–1114. [Google Scholar] [CrossRef]
- Pavone, P.; Marino, S.; Maniaci, A.; Cocuzza, S. Aarskog-Scott syndrome: Clinical and molecular characterisation of a family with the coexistence of a novel FGD1 mutation and 16p13.11-p12.3 microduplication. BMJ Case Rep. 2020, 13, e235183. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Coria, M.; Lugo-Trampe, J.J.; Zamudio-Osuna, M.; Rodríguez-Sánchez, I.P.; Lugo-Trampe, A.; de la Fuente-Cortez, B.; Campos-Acevedo, L.D.; Martínez-de-Villarreal, L.E. Identification of novel mutations in Mexican patients with Aarskog-Scott syndrome. Mol. Genet. Genom. Med. 2015, 3, 197–202. [Google Scholar] [CrossRef]
- Pizio, H.F.; Scott, M.H.; Richard, J.M. Tortuosity of the retinal vessels in Aarskog syndrome (faciogenital dysplasia). Ophthalmic Genet. 1994, 15, 37–40. [Google Scholar] [CrossRef]
- Sariyilmaz, K.; Ozkunt, O.; Korkmaz, M.; Dikici, F.; Domanic, U. Aarskog-Scott syndrome: An unusual cause of scoliosis. J. Craniovertebral Junction Spine 2017, 8, 283–284. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Gillessen-Kaesbach, G.; May, M.; Cappa, M.; Gorski, J.; Steindl, K.; Neri, G. Two novel mutations confirm FGD1 is responsible for the Aarskog syndrome. Eur. J. Hum. Genet. 2000, 8, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Shalev, S.A.; Chervinski, E.; Weiner, E.; Mazor, G.; Friez, M.J.; Schwartz, C.E. Clinical variation of Aarskog syndrome in a large family with 2189delA in the FGD1 gene. Am. J. Med. Genet. A 2006, 140, 162–165. [Google Scholar] [CrossRef]
- Shinkawa, T.; Yamauchi, Y.; Osada, Y.; Ishisawa, N. Aarskog syndrome. Urology 1983, 22, 624–626. [Google Scholar] [CrossRef]
- Stevenson, R.E.; May, M.; Arena, J.F.; Millar, E.A.; Scott, C.I., Jr.; Schroer, R.J.; Simensen, R.J.; Lubs, H.A.; Schwartz, C.E. Aarskog-Scott syndrome: Confirmation of linkage to the pericentromeric region of the X chromosome. Am. J. Med. Genet. 1994, 52, 339–345. [Google Scholar] [CrossRef]
- Takahashi, I.; Noguchi, A.; Kondo, D.; Sato, Y.; Suzuki, H.; Yamada, M.; Kosaki, K.; Takahashi, T. A novel missense variant of FGD1 disrupts critical cysteine residues of the FYVE domain in Japanese siblings with Aarskog-Scott syndrome. Clin. Pediatr. Endocrinol. 2024, 33, 39–42. [Google Scholar] [CrossRef]
- Tsukahara, M.; Fernandez, G.I. Umbilical findings in Aarskog syndrome. Clin. Genet. 1994, 45, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Völter, C.; Martínez, R.; Hagen, R.; Kress, W. Aarskog-Scott syndrome: A novel mutation in the FGD1 gene associated with severe craniofacial dysplasia. Eur. J. Pediatr. 2014, 173, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Vooren, M.J.; Niermeijer, M.F.; Hoogeboom, A.J. The Aarskog syndrome in a large family, suggestive for autosomal dominant inheritance. Clin. Genet. 1983, 24, 439–445. [Google Scholar] [CrossRef]
- Zielinski, J.A.; Pack, L.L. Bilateral anterior hip dislocation in a child with Aarskog syndrome: A case report. J. Pediatr. Orthop. 2008, 28, 729–732. [Google Scholar] [CrossRef]
- Hyman, S.L.; Levy, S.E.; Myers, S.M.; Council on Children with Disabilities, Section on Developmental and Behavioral Pediatrics. Identification, Evaluation, and Management of Children with Autism Spectrum Disorder. Pediatrics 2020, 145, e20193447. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanova, R.S.; Talantseva, O.I.; Lind, K.V.; Manasevich, V.A.; Kuznetsova, J.E.; Grigorenko, E.L. The Co-Occurrence of Autism Spectrum Disorder and Aarskog–Scott Syndrome in an Accomplished Young Man. Pediatr. Rep. 2025, 17, 73. https://doi.org/10.3390/pediatric17040073
Romanova RS, Talantseva OI, Lind KV, Manasevich VA, Kuznetsova JE, Grigorenko EL. The Co-Occurrence of Autism Spectrum Disorder and Aarskog–Scott Syndrome in an Accomplished Young Man. Pediatric Reports. 2025; 17(4):73. https://doi.org/10.3390/pediatric17040073
Chicago/Turabian StyleRomanova, Raisa S., Oksana I. Talantseva, Katerina V. Lind, Victoria A. Manasevich, Julia E. Kuznetsova, and Elena L. Grigorenko. 2025. "The Co-Occurrence of Autism Spectrum Disorder and Aarskog–Scott Syndrome in an Accomplished Young Man" Pediatric Reports 17, no. 4: 73. https://doi.org/10.3390/pediatric17040073
APA StyleRomanova, R. S., Talantseva, O. I., Lind, K. V., Manasevich, V. A., Kuznetsova, J. E., & Grigorenko, E. L. (2025). The Co-Occurrence of Autism Spectrum Disorder and Aarskog–Scott Syndrome in an Accomplished Young Man. Pediatric Reports, 17(4), 73. https://doi.org/10.3390/pediatric17040073